
التهاب الكلية الوراثي (متلازمة ألبورت) عند الأطفال
آخر مراجعة: 23.04.2024

تتم مراجعة جميع محتويات iLive طبياً أو التحقق من حقيقة الأمر لضمان أكبر قدر ممكن من الدقة الواقعية.
لدينا إرشادات صارمة من مصادرنا ونربط فقط بمواقع الوسائط ذات السمعة الطيبة ، ومؤسسات البحوث الأكاديمية ، وطبياً ، كلما أمكن ذلك استعراض الأقران الدراسات. لاحظ أن الأرقام الموجودة بين قوسين ([1] و [2] وما إلى ذلك) هي روابط قابلة للنقر على هذه الدراسات.
إذا كنت تشعر أن أيًا من المحتوى لدينا غير دقيق أو قديم. خلاف ذلك مشكوك فيه ، يرجى تحديده واضغط على Ctrl + Enter.
التهاب الكلية وراثي (داء ألبورت) - قررت وراثيا غير المناعة الموروثة اعتلال كبيبات الكلى المعرض البيلة الدموية (في بعض الأحيان بروتينية)، وانخفاض تدريجي في وظائف الكلى المزمنة في تطوير الفشل الكلوي وكثيرا ما يرتبط مع الصمم الحسي العصبي وضعاف البصر.
لأول مرة تم وصف المرض في عام 1902 من قبل LGGuthrie ، الذي لاحظ وجود عائلة في عدة أجيال والتي لوحظ بيلة دموية. في عام 1915 ، وصف أفراد من عائلة AFHurst نفسها تطور يوريمية. في عام 1927 ، حددت Al Alport لأول مرة الصمم في العديد من الأقارب مع بيلة دموية.في 50s من القرن الماضي ، تم وصف إصابات العين في مثل هذا المرض. في عام 1972 ، في المرضى الذين يعانون من بيلة دموية وراثية ، وفحص شكلي الأنسجة الكلى ، هينجليس وآخرون. كشف توسع غير متكافئ وتلطيخ الأغشية القاعدية الكبيبية. في عام 1985 ، تم تحديد الأساس الجيني للالتهاب الكبدي الوراثي - وهو طفرة في جين نوع الكولاجين الرابع (فينغولد وآخرون ، 1985).
أدى التحقيق في الطبيعة الوراثية للمرض إلى استنتاج أن الاختلافات في مظاهر المظاهر الظاهرية للالتهاب الكبدي الوراثي (مع أو بدون فقدان السمع) ترجع إلى درجة التعبير عن الجين الطافر. وبالتالي ، في الوقت الحاضر تعتبر جميع المتغيرات السريرية مظاهر للمرض واحد ومصطلح "التهاب الكلية الوراثي" هو مرادف لمصطلح "متلازمة آلبورت".
ووفقاً للدراسات الوبائية ، يحدث التهاب الكلية الوراثي بمعدل 17 لكل 100،000 طفل.
أسباب متلازمة ألبورت
الأساس الجيني للمرض هو طفرة في الجين أ 5 من سلسلة الكولاجين من النوع الرابع. هذا النوع من عالمي لأغشية القاعدية الكلى، جهاز القوقعة، وكبسولات العدسة، وشبكية العين والقرنية هو موضح في الدراسات باستخدام الأجسام المضادة وحيدة النسيلة ضد الكسر الكولاجين. في الآونة الأخيرة ، فإنها تشير إلى إمكانية استخدام تحقيقات الحمض النووي لتشخيص ما قبل الولادة من التهاب الكلية الوراثي.
يتم التأكيد على أهمية اختبار جميع أفراد الأسرة باستخدام تحقيقات الحمض النووي لتحديد ناقلات الجين الطافر ، وهو أمر ذو أهمية كبيرة في إجراء الاستشارة الوراثية الطبية للأسر التي تعاني من هذا المرض. ومع ذلك ، فإن ما يصل إلى 20٪ من العائلات ليس لديها أقارب يعانون من أمراض الكلى ، وهو ما يشير إلى ارتفاع معدل حدوث طفرات عفوية في الجين غير الطبيعي. غالبية المرضى الذين يعانون من التهاب الكلى الوراثي في الأسر لديهم أفراد يعانون من أمراض الكلى وفقدان السمع وأمراض الرؤية. الزواج ذو الصلة بين الأشخاص الذين لديهم واحد أو أكثر من الأسلاف ، لأن زواج الأفراد المرتبطين به يزيد من احتمال الحصول على نفس الجينات من كلا الوالدين. يتم تأسيس صبغية سائدة وراثي جسمي متنحي ومسيطر ، مرتبطة بالكروموسوم X لمسار الإرسال.
من الأرجح أن يميز الأطفال ثلاثة أنواع مختلفة من التهاب الكلية الوراثي: متلازمة ألبورت ، والتهاب الكلوي الوراثي دون فقدان السمع ، وبيلة دموية حميدة للأسرة.
متلازمة آلبورت - التهاب الكلية الوراثي مع تلف السمع. الأساس هو خلل مشترك في بنية الكولاجين للغشاء القاعدي للكبيبات في الكليتين ، بنية الأذن والعين. يقع جين متلازمة آلبورت الكلاسيكية في موقع 21-22 q من الذراع الطويلة للكروموسوم X. في معظم الحالات ، يتم توريثه من النوع المسيطر المرتبط بالكروموسوم X. في هذا الصدد ، في الرجال ، تكون متلازمة ألبورت أكثر صعوبة ، لأنه في النساء يتم تعويض وظيفة الجين الطافر بواسطة أليل صحي للكروموسوم الثاني ، السليم.
الأساس الوراثي لتطور التهاب الكلية الوراثي هي طفرات في جينات سلاسل ألفا من نوع الكولاجين الرابع. كما هو معروف ستة سلاسل من نوع IV الكولاجين G: A5 و A6 جينات سلاسل (Sol4A5 وSol4A5) تقع على الذراع الطويلة للكروموسوم X في منطقة 21-22q. جينات سلاسل a3 و a4 (Co4A3 و Co4A4) - على الكروموسوم الثاني جينات سلاسل a1 و a2 (Co4A1 و Co4A2) - على الكروموسوم الثالث عشر.
في معظم الحالات (80-85 ٪) ، يرتبط نوع من الوراثة المرتبطة بالـ X مع تلف في الجين Co4A5 بسبب الحذف أو الطفرات النقطية أو اضطرابات الربط. حاليا ، تم العثور على أكثر من 200 طفرة من الجينات Kol4A5 ، المسؤولة عن انتهاك تخليق سلسلة 5 من الكولاجين الرابع. في هذا النوع من الميراث ، يتجلى المرض في الأطفال من كلا الجنسين ، ولكن في الأولاد يكون أكثر صعوبة.
ورثت الطفرات في موقع الجينات Co4A3 و Co4A4 ، المسؤولة عن تخليق a3 و a4 - سلاسل من نوع الكولاجين الرابع ، بشكل وراثي. وفقا لبحث ، لوحظ وجود نوع وراثي جسمي قاهر من الميراث في 16 ٪ من حالات التهاب الكلية الوراثي ، وراثي جسمي مقهور - في 6 ٪ من المرضى. هناك حوالي 10 طفرات من جينات Co4A3 و Co4A4.
نتيجة الطفرات هي انتهاك لعمليات التجميع من نوع الكولاجين الرابع ، مما يؤدي إلى خلل في هيكلها. يعتبر نوع الكولاجين الرابع أحد المكونات الرئيسية للغشاء القاعدي الكبيبي ، وجهاز القوقعة وعدسة العين ، وسيتم الكشف عن علم الأمراض في عيادة التهاب الكلية الوراثي.
نوع الكولاجين IV، وهي جزء من الغشاء القاعدي الكبيبي، ويتكون أساسا من سلسلتين A1 (IV) وسلسلة A2 واحد (IV)، ويحتوي أيضا على A3، A4، A5 سلسلة. في أغلب الأحيان عندما الميراث Sol4A5 طفرة مرتبطة X يرافقه نقص A3، A4 أو A5 A6 وسلاسل من نوع الكولاجين الرابع في الهيكل، وعدد من O1 وسلاسل A2 إلى الزيادات الغشاء القاعدي الكبيبي. آلية هذه الظاهرة غير واضحة ، ويفترض أن السبب هو التغييرات posttranscriptional في مرنا.
A3 نقص، A4 أو وسلاسل A5 في نوع هيكل غشاء IV الكولاجين الطابق السفلي من نتائج الكبيبات في ترقق وهشاشة المراحل المبكرة من متلازمة آلبورت أن يتجلى سريريا معظم البيلة الدموية (أحيانا بيلة دموية أو بروتينية فقط بروتينية)، وفقدان ومخروط العدسة السمع. مزيد من تطور المرض يؤدي إلى سماكة، وتعطيل نفاذية الغشاء القاعدي في مراحل متأخرة من المرض، مع نمو في هذه الأنواع الكولاجين الخامس والسادس، والذي تجلى في زيادة بروتينية وانخفاض وظائف الكلى.
طبيعة الطفرة الكامنة وراء التهاب الكلية الوراثي يحدد إلى حد كبير مظهر المظهري. عندما X الحذف كروموسوم مع الطفرة في وقت واحد والجينات Sol4A6 Sol4A5 المسؤولة عن تركيب A5 و A6 سلاسل من نوع الكولاجين IV، جنبا إلى جنب مع داء ألبورت المريء ورام عضلي أملس والأعضاء التناسلية. ووفقا للدراسات مع الطفرات الجينية Sol4A5 المرتبطة الحذف يتم وضع علامة شدة كبيرة من عملية المرضية، وهو مزيج مع آفة الكلى مظاهر خارج الكلية والتنمية في وقت مبكر من الفشل الكلوي المزمن، مقارنة طفرة stochechnoy من هذا الجين.
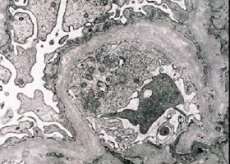
من الناحية المورفولوجية ، يكشف الفحص المجهري الإلكتروني عن ترقيق وتفتيت الأغشية القاعدية الكبيبية (خاصة الصفيحة densa) ووجود حبيبات كثيفة إلكترونياً. قد تكون آفة الكبيبة غير متماثلة في نفس المريض ، من الحد الأدنى من الآفة البؤرية للسان العموم إلى الكبيبات. التهاب كبيبات الكلى في متلازمة ألبورت هو دائمًا سلبي المناعة ، والذي يميزه عن التهاب كبيبات الكلى. سمة هي تطوير ضمور القناة ، تسلل lymphohistiocyte ، وجود "خلايا رغوة" مع شوائب من الدهون - lipofagi. مع تطور المرض ، تم الكشف عن تدمير سميك وتميز ملحوظ في أغشية كبيبات القاعدية.
يتم الكشف عن بعض التغييرات في حالة الجهاز المناعي. المرضى الذين يعانون من التهاب الكلية وراثي انخفض مستوى الإيج A، والميل إلى زيادة في تركيز الدم من الغلوبولين المناعي، ويمكن زيادة مستوى مفتش في المراحل المبكرة من المرض وانخفاض في مراحل لاحقة. ربما تكون الزيادة في تركيز IgM و G هي نوع من الاستجابة التعويضية استجابة لعجز IgA.
يتم تقليل النشاط الوظيفي لنظام T اللمفاويات. انه يمثل الحد الانتقائي للB-الخلايا الليمفاوية، المسؤولة عن تركيب الإيج A، كسر الارتباط الحصانة أكلة، ويرجع ذلك أساسا إلى انهيار الكيميائي العمليات والهضم داخل الخلايا من العدلات
في دراسة خزعة الكلى في المرضى الذين يعانون من متلازمة آلبورت بواسطة المجهر الإلكتروني، والتغيرات التركيبية لاحظت الكبيبي الطابق السفلي غشاء: رقيق، وأنماط تقسيم انتهاك غشاء قاعدي الكبيبي مع التغير في سمك ومعالم متفاوتة. في المراحل المبكرة من التهاب الكلية الوراثي ، يحدد العيب ترقق وهشاشة الأغشية القاعدية الكبيبية.
ترقق الأغشية الكبيبي هو علامة أكثر ملاءمة وأكثر شيوعا في الفتيات. هناك ميزة أكثر إلكترونًا في الميكروبات في التهاب الكلية الوراثي هي انقسام الغشاء القاعدي ، وتترافق شدة دماره مع شدة العملية.

أعراض متلازمة ألبورت عند الأطفال
تظهر الأعراض الأولى لمتلازمة ألبورت في شكل متلازمة بولية معزولة في الأطفال في السنوات الثلاث الأولى من الحياة. في معظم الحالات ، يتم الكشف عن المرض عن طريق الصدفة. يتم الكشف عن المتلازمة البولية أثناء الفحص الوقائي للطفل ، قبل الدخول إلى مؤسسة الأطفال أو خلال ARVI. في حالة ظهور أمراض في البول خلال ARVI. في التهاب الكلية الوراثي ، على عكس التهاب كبيبات الكلى المكتسب ، لا توجد فترة كامنة.
في المرحلة الأولى من المرض ، يعاني رفاه الطفل قليلاً ، والميزة المميزة هي استمرار واستمرار المتلازمة البولية. واحدة من العلامات الرئيسية هي بيلة دموية بدرجات متفاوتة ، لوحظت في 100 ٪ من الحالات. ويلاحظ زيادة في درجة بيلة دموية أثناء أو بعد التهابات الجهاز التنفسي ، والمجهود البدني أو بعد التطعيمات الوقائية. البروتين في معظم الحالات لا يتجاوز 1 غرام / يوم ، في بداية المرض قد يكون غير مستقر ، مع تقدم عملية زيادة بروتينية. بشكل دوري ، يمكن أن يكون للرواسب البولية الكريات البيض مع غلبة الخلايا الليمفاوية ، والتي ترتبط مع تطور التغيرات الخلالي.
في وقت لاحق ، هناك انتهاك للوظائف الجزئية للكلية ، وتفاقم الحالة العامة للمريض: التسمم ، وضعف العضلات ، انخفاض ضغط الدم الشرياني ، غالباً ضعف السمع (خاصة عند الأولاد) ، ضعف البصر في بعض الأحيان. يظهر التسمم كالشحوب ، والتعب ، والصداع. في المرحلة الأولى من المرض ، يتم الكشف عن فقدان السمع في معظم الحالات فقط عن طريق التصوير السمعي. يمكن أن يحدث فقدان السمع في متلازمة ألبورت في فترات مختلفة من مرحلة الطفولة ، ولكن في معظم الأحيان يتم تشخيص فقدان السمع في سن 6-10 سنوات. يبدأ فقدان السمع عند الأطفال عند ترددات عالية ، ويصل إلى درجة كبيرة في الهواء والتوصيل العظمي ، ويمرّ من الصوت إلى الصمّ المتلقي للصوت. يمكن أن يكون فقدان السمع أحد الأعراض الأولى للمرض ويمكن أن يسبق متلازمة البولية.
في 20 ٪ من الحالات ، يعاني مرضى متلازمة ألبورت من تغيرات في العين. الشذوذ الأكثر شيوعا من العدسة: spherofokiya ، lentikonus الأمامية والخلفية أو مختلطة ، ومجموعة متنوعة من إعتام عدسة العين. في العائلات التي تعاني من متلازمة آلبورت ، هناك عدد كبير من حالات قصر النظر. لاحظ عدد من الباحثين باستمرار في هذه العائلات تغيرات ثنائية في شكل تحبيب براق أو أصفر مائل للصفرة في منطقة الجسم الأصفر. يعتبرون هذا العرض بمثابة عرض دائم ، والذي له قيمة تشخيصية عالية في متلازمة ألبورت. C. S. Chugh et al. (1993) لدراسة طب العيون كشف آلبورت مرضى متلازمة نقص حدة الإبصار في 66.7٪ من الحالات، ومخروط العدسة إلى الأمام - 37.8٪، والبقع على شبكية العين - في 22،2٪، وإعتام عدسة العين - 20٪، القرنية المخروطية - 6 7٪
في بعض الأطفال المصابين بالتهاب الكلية الوراثي ، وخاصة في تشكيل القصور الكلوي ، لوحظ تأخر كبير في النمو الجسدي. كما تقدم تطور القصور الكلوي ارتفاع ضغط الدم. في الأطفال ، يتم الكشف عنها في كثير من الأحيان في مرحلة المراهقة وفي الفئات العمرية الأكبر سنا.
مميزة هو وجود في المرضى الذين يعانون من التهاب الكلية الوراثي من الوصمات (أكثر من 5-7) مختلفة من dysembryogenesis النسيج الضام. بين النسيج الضام من وصمة العار في المرضى الذين يعانون من فرط الأكثر شيوعا العين والحنك عالية، سوء الإطباق، شكل غير طبيعي من الأذنين، وانحناء الإصبع الصغير على يديه، "فجوة sandalevidnaya" على القدمين. يتميز التهاب الكلية الوراثي بنفس النوع من التسمم الناتج عن دماغ الثدي داخل الأسرة ، فضلاً عن ارتفاع تواتر انتشاره بين أقارب المحلل ، الذي ينتقل المرض من خلاله.
في المراحل المبكرة للمرض كشف تخفيض معزولة وظائف الكلى جزئية: نقل الأحماض الأمينية، والشوارد، وظائف تركيز Acidogenesis، المزيد من التغييرات حالة وظيفية على حد سواء القريبة والبعيدة كليون ولها طابع اضطرابات جزئية مجتمعة. الحد من الترشيح الكبيبي يحدث في وقت لاحق ، في كثير من الأحيان في فترة المراهقة. مع تقدم التهاب الكلية الوراثي ، تطور فقر الدم.
وهكذا، على راثي التهاب الكلية انطلاق يتميز المرض: المرحلة الأولى كامنة أو الأعراض السريرية الخفية التي اظهرها تغيرات طفيفة متلازمة المثانة ثم تحدث عملية تدريجية المعاوضة مع انخفاض في وظائف الكلى مع أعراض سريرية العلنية (التسمم، والوهن، وتأخر في النمو، anemizatsiya). تظهر الأعراض السريرية عادة بغض النظر عن التقسيم الطبقي للتفاعل الالتهابي.
يمكن أن يظهر التهاب الكلية الوراثي في الفترات العمرية المختلفة ، والتي تعتمد على عمل الجين ، والذي يكون حتى وقت معين في حالة مكبوتة.
تصنيف
هناك ثلاثة أنواع من التهاب الكلية الوراثي
- أنا البديل - يتجلى سريريا التهاب الكلية مع بيلة دموية ، وفقدان السمع وتلف العين. مسار التهاب الكلية هو تقدمية مع تطور CRF. نوع الميراث هو المسيطر ، المرتبط بالكروموسوم X. من الناحية المورفولوجية ، هناك اضطراب في بنية الغشاء القاعدي ، وترققه وانشقاقه.
- يتجلى II البديل - سريريا التهاب الكلية مع بيلة دموية دون فقدان السمع. مسار التهاب الكلية هو تقدمية مع تطور الفشل الكلوي المزمن. نوع الميراث هو المسيطر ، المرتبط بالكروموسوم X. مورفولوجياً ، تم الكشف عن ترقق الغشاء القاعدي للشعيرات الكبيبية (خاصة laminadensa).
- الخيار الثالث - بيلة دموية الأسرة الحميدة. الدورة مواتية ، الفشل الكلوي المزمن لا يتطور. نوع الميراث هو وراثي جسمي قاهر أو مقهورة. في نوع وراثي جسمي متنحي من الميراث ، يكون لدى النساء مسار أكثر شدة للمرض.
تشخيص متلازمة ألبورت
يتم اقتراح المعايير التالية:
- وجود في كل عائلة من مريضين على الأقل مع اعتلال الكلية.
- بيلة دموية باعتبارها العرض الرئيسي لاعتلال الكلية في proband.
- يعاني فرد واحد على الأقل من أفراد العائلة من فقدان السمع ؛
- تطوير الفشل الكلوي المزمن في واحد قريب وأكثر.
في تشخيص مجموعة متنوعة من الأمراض الوراثية والخلقية مكانا هاما ينتمي إلى نهج متكامل للتفتيش وقبل كل شيء الالتفات إلى البيانات التي تم الحصول عليها في إعداد نسب الطفل. تعتبر تشخيص متلازمة آلبورت صالح في الحالات التي يكون فيها المريض 3 من 4 المظاهر التقليدية: وجود في بيلة دموية الأسرة والفشل الكلوي المزمن، وجود فقدان السمع الحسي العصبي للمريض، وأمراض الكشف في الإلكترون علامات مجهرية توصيف خزعة الشق الغشاء القاعدي الكبيبي مع تغيير سمك وخطوط متفاوتة.
يجب أن يتضمن فحص المريض الطرق الجينية السريرية للتحقيق. دراسة موجهة من سوابق المرض. الفحص العام للمريض مع الأخذ بعين الاعتبار معايير التشخيص. في مرحلة التعويض ، يمكن للمرء أن يمسك علم الأمراض فقط من خلال التركيز على مثل هذه المتلازمات مثل وجود مضاعفات وراثية ، انخفاض ضغط الدم ، وصمة عار متعددة من خلل تنسج الدهون ، وتغيرات في متلازمة البولية. في مرحلة انهيار المعاوضة ، قد يكون هناك ظهور لأعراض estrarenal ، مثل التسمم الحاد ، والوهن ، متخلفة في النمو البدني ، وفقر الدم ، ويتجلى ويتضخم مع انخفاض تدريجي في وظيفة الكلى. في معظم المرضى الذين يعانون من انخفاض في وظيفة الكلى ، لوحظ انخفاض في وظيفة من acido- و aminogenesis. في 50 ٪ من المرضى لاحظ انخفاض كبير في وظيفة إفرازية من الكلى. الحد من مدى التقلبات في الكثافة البصرية للبول ؛ انتهاكا لإيقاع الترشيح ، ثم انخفاض في الترشيح الكبيبي. يتم تشخيص مرحلة الفشل الكلوي المزمن لدى المرضى الذين يعانون من مستوى اليوريا لمدة 3-6 أشهر وأكثر ارتفاعًا في مصل الدم (أكثر من 0.35 جم / لتر) ، انخفاض في الترشيح الكبيبي إلى 25٪ من القاعدة.
يجب إجراء التشخيص التفريقي للالتهاب الكبدي الوراثي في المقام الأول مع شكل البكتريا من التهاب كبيبات الكلى المكتسب. اكتسبت التهاب كبيبات الكلى الحاد على نحو متزايد بداية فترة 2-3 أسابيع بعد الإصابة السابقة، وميزات خارج الكلية، بما في ذلك ارتفاع ضغط الدم مع الأيام الأولى (في التهاب الكلية وراثية، على العكس، انخفاض ضغط الدم)، وانخفض معدل الترشيح الكبيبي في البداية، أي انتهاك للوظائف أنبوبي جزئية، في حين كما هو الحال مع الوراثي فهي موجودة. يحدث التهاب كبيبات الكلى المكتسبة مع بيلة دموية وبروتينية أكثر وضوحًا ، مع زيادة ESR. إن التغيرات النموذجية في الغشاء القاعدي الكبيبي الذي يتميز به التهاب الكلية الوراثي هي ذات أهمية تشخيصية.
يتم إجراء التشخيص التفريقي لاعتلال الكلية الذي يعاني من خلل وظيفي مع الفشل الكلوي المزمن ، وتعرف الأسرة سريريا أنواع متعددة من أمراض الكلى ، وربما طيف من اعتلال الكلية من التهاب الحويضة والكلية إلى التحصين. الأطفال غالبا ما يكون لديهم شكاوى من آلام في البطن وبشكل دوري مع التبول ، في رواسب البول - أكسالات.
إذا كنت تشك في التهاب الكلية الوراثي يجب إرسال المريض لتوضيح التشخيص في قسم أمراض الكلى المتخصصة.
ما الذي يجب فحصه؟
ما هي الاختبارات المطلوبة؟
من الاتصال؟
علاج متلازمة ألبورت
في النظام ينص على تقييد المجهود البدني الكبير ، والبقاء في الهواء النقي. النظام الغذائي هو عالي الجودة ، مع محتوى كاف من البروتينات والدهون والكربوهيدرات عالية الجودة مع الأخذ بعين الاعتبار وظيفة الكليتين. من أهمية كبيرة هو تحديد وإعادة تأهيل بؤر العدوى المزمنة. من الأدوية ، ATP ، cocarboxylase ، البيريدوكسين (تصل إلى 50 ملغ / يوم) ، ويستخدم كلوريد الكارنيتين. تعقد الدورات 2-3 مرات في السنة. عندما يتم وصف بيلة دموية العلاج النباتي - نبات القراص ، نبات القراص ، الرماد بلاك ، يارو.
في الأدب الأجنبي والمحلي هناك تقارير عن العلاج بالبريدنيزولون واستخدام التخلاء. ومع ذلك ، من الصعب الحكم على التأثير.
في الفشل الكلوي المزمن ، يتم استخدام غسيل الكلى وزرع الكلى.
لا توجد طرق لعلاج محدد (إمراضي فعال) من التهاب الكلية الوراثي. تهدف جميع التدابير الطبية إلى منع وإبطاء الحد من وظائف الكلى.
يجب أن يكون النظام الغذائي متوازنا وذات سعرات حرارية عالية ، مع مراعاة الحالة الوظيفية للكلية. في حالة عدم وجود انتهاكات للحالة وظيفية في تغذية الطفل يجب أن يكون محتوى كاف من البروتينات والدهون والكربوهيدرات. في وجود علامات على الفشل الكلوي ، يجب أن تكون كمية البروتين والكربوهيدرات من الكالسيوم والفوسفور محدودة ، مما يؤخر تطور الفشل الكلوي المزمن.
يجب أن يكون الإجهاد البدني محدودًا ، وينصح الأطفال بالامتناع عن ممارسة الرياضة.
تجنب الاتصال مع المرضى المصابين بالعدوى ، والحد من مخاطر الإصابة بالتهابات الجهاز التنفسي الحادة. من الضروري تعقيم بؤر العدوى المزمنة. لا يتم إجراء التطعيمات الوقائية للأطفال المصابين بالتهاب الكلية الوراثي ، ولا يمكن التطعيم إلا وفقاً للإشارات الوبائية.
العلاج الهرموني والمثبط في التهاب الكلية الوراثي غير فعال. هناك مؤشرات على وجود تأثير إيجابي معين (انخفاض في مستوى بروتينية وبطء في تطور المرض) مع استخدام مثبطات السيكلوسبورين A و ACE على المدى الطويل لسنوات عديدة.
في علاج المرضى الذين يستخدمون الأدوية التي تعمل على تحسين عملية الأيض:
- البيريدوكسين - 2-3 ملغم / كغم / يوم في 3 جرعات مقسمة لمدة 4 أسابيع ؛
- kokarboksilaza - 50 ملغ في العضل كل يوم ، فقط 10-15 حقنة ؛
- اعبي التنس المحترفين - 1 مل في العضل كل يوم ، 10-15 الحقن.
- فيتامين أ - 1000 وحدة في السنة / يوم في حفل استقبال لمدة أسبوعين ؛
- فيتامين هـ - 1 مغ / كغ / يوم في استقبال واحد لمدة أسبوعين.
هذا العلاج يحسن الحالة العامة للمرضى ، ويقلل من الخلل الأنبوبي ، ويتم إدارته 3 مرات في السنة.
كما يمكن استخدام immunomodulator - 2 ملغم / كغ / يوم 2-3 مرات في الأسبوع مع intermissions بين جرعات 3-4 أيام.
للباحثين ، الأوكسجين الضغط العالي له تأثير إيجابي على شدة بيلة دموية وخلل كلوي.
الطريقة الأكثر فعالية لعلاج التهاب الكلية الوراثي هي زرع الكلى في الوقت المناسب. لا يوجد تكرار للمرض في الزرع ، في نسبة صغيرة من الحالات (حوالي 5 ٪) ، من الممكن تطوير التهاب الكلية في الكلي المزروع المرتبط بالمستضدات إلى الغشاء القاعدي الكبيبي.
منطقة واعدة هي التشخيص قبل الولادة والعلاج والهندسة الوراثية. تظهر التجارب على الحيوانات كفاءة عالية في نقل جينات طبيعية مسؤولة عن تركيب سلاسل من نوع الكولاجين الرابع لنسيج الكلى ، وبعد ذلك يتم ملاحظة تخليق هياكل الكولاجين الطبيعية.
توقعات
إن التشخيص من التهاب الكلية الوراثي هو دائما جاد.
المعايير غير المواتية للإحباط لتدفق التهاب الكلية الوراثي هي:
- الجنس الذكور
- التطور المبكر للفشل الكلوي المزمن في أفراد الأسرة ؛
- بروتينية (أكثر من 1 غم / يوم) ؛
- سماكة الأغشية القاعدية الكبيبي وفقا للفحص المجهري ؛
- التهاب العصب العصبي السمعي.
- الحذف في الجين Co4A5.
إن تشخيص بيلة دموية أسرية حميدة أكثر ملاءمة.
Использованная литература