خبير طبي في المقال

منشورات جديدة
التهاب الكلية الوراثي (متلازمة ألبورت) لدى الأطفال
Last reviewed: 05.07.2025

تتم مراجعة جميع محتويات iLive طبياً أو التحقق من حقيقة الأمر لضمان أكبر قدر ممكن من الدقة الواقعية.
لدينا إرشادات صارمة من مصادرنا ونربط فقط بمواقع الوسائط ذات السمعة الطيبة ، ومؤسسات البحوث الأكاديمية ، وطبياً ، كلما أمكن ذلك استعراض الأقران الدراسات. لاحظ أن الأرقام الموجودة بين قوسين ([1] و [2] وما إلى ذلك) هي روابط قابلة للنقر على هذه الدراسات.
إذا كنت تشعر أن أيًا من المحتوى لدينا غير دقيق أو قديم. خلاف ذلك مشكوك فيه ، يرجى تحديده واضغط على Ctrl + Enter.
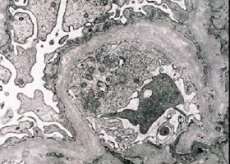
التهاب الكلية الوراثي (متلازمة ألبورت) هو اعتلال كبيبات الكلى الوراثي غير المناعي المحدد وراثيًا، يتجلى في وجود دم في البول (أحيانًا مع بروتين في البول)، والتدهور التدريجي في وظائف الكلى مع تطور الفشل الكلوي المزمن، وغالبًا ما يقترن بالصمم الحسي العصبي وضعف البصر.
وُصف هذا المرض لأول مرة عام ١٩٠٢ من قِبل إل جي جوثري، الذي لاحظ وجود بيلة دموية في عائلة على مدى عدة أجيال. وفي عام ١٩١٥، وصف إيه إف هيرست تطور داء بولينا لدى أفراد من نفس العائلة. وفي عام ١٩٢٧، حدّد إيه. ألبورت لأول مرة فقدان السمع لدى العديد من الأقارب المصابين ببيلة دموية. وفي خمسينيات القرن الماضي، وُصفت آفات العين المرتبطة بمرض مماثل. وفي عام ١٩٧٢، كشف هينجلايس وآخرون، خلال دراسة مورفولوجية لأنسجة الكلى لدى مرضى يعانون من بيلة دموية وراثية، عن تمدد وطبقية غير متساوية في الأغشية القاعدية الكبيبية. وفي عام ١٩٨٥، حُدد الأساس الجيني لالتهاب الكلية الوراثي - وهو طفرة في جين الكولاجين من النوع الرابع (فينجولد وآخرون، ١٩٨٥).
أتاحت لنا دراسة الطبيعة الجينية للمرض استنتاج أن الاختلافات في المظاهر الظاهرية لالتهاب الكلية الوراثي (سواءً مع فقدان السمع أو بدونه) ترجع إلى درجة التعبير عن الجين الطافر. لذا، تُعتبر جميع المتغيرات السريرية حاليًا مظاهر لمرض واحد، ويُعتبر مصطلح "التهاب الكلية الوراثي" مرادفًا لمصطلح "متلازمة ألبورت".
وبحسب الدراسات الوبائية فإن التهاب الكلية الوراثي يحدث بمعدل 17 حالة لكل 100 ألف طفل.
 [
[ أسباب متلازمة ألبورت
الأساس الجيني للمرض هو طفرة في جين السلسلة a-5 من الكولاجين من النوع الرابع. هذا النوع شائع في الأغشية القاعدية للكلى، والقوقعة، وكبسولة العدسة، وشبكية العين، وقرنية العين، وقد ثبت ذلك في دراسات أجريت باستخدام أجسام مضادة وحيدة النسيلة ضد هذا الجزء من الكولاجين. ومؤخرًا، أُشير إلى إمكانية استخدام مسابر الحمض النووي لتشخيص التهاب الكلية الوراثي قبل الولادة.
يُشدد على أهمية فحص جميع أفراد العائلة باستخدام مسابر الحمض النووي (DNA) لتحديد حاملي الجين الطافر، وهو أمر بالغ الأهمية في تقديم الاستشارات الطبية والوراثية للعائلات المصابة بهذا المرض. ومع ذلك، فإن ما يصل إلى 20% من العائلات ليس لديها أقارب يعانون من أمراض الكلى، مما يشير إلى ارتفاع وتيرة الطفرات التلقائية للجين الشاذ. لدى معظم مرضى التهاب الكلية الوراثي أفراد يعانون من أمراض الكلى وفقدان السمع وأمراض الرؤية في عائلاتهم؛ كما أن زواج الأقارب بين الأشخاص الذين لديهم سلف واحد أو أكثر أمر مهم، حيث يزداد احتمال تلقي نفس الجينات من كلا الوالدين في زواج الأقارب. وقد تم تحديد طرق انتقال المرض، وهي السائدة، والمتنحية، والمرتبطة بالكروموسوم X.
عند الأطفال، يتم التمييز بين ثلاثة أنواع من التهاب الكلية الوراثي بشكل شائع: متلازمة ألبورت، والتهاب الكلية الوراثي دون فقدان السمع، والبيلة الدموية الحميدة العائلية.
متلازمة ألبورت هي التهاب كلوي وراثي مصحوب بضعف سمع. وتستند إلى خلل مشترك في بنية الكولاجين في الغشاء القاعدي الكبيبي للكلى والأذن والعين. يقع جين متلازمة ألبورت الكلاسيكية في الموضع 21-22q من الذراع الطويلة للكروموسوم X. في معظم الحالات، يُورث هذا الجين بشكل سائد، مرتبطًا بالكروموسوم X. في هذا الصدد، تكون متلازمة ألبورت أكثر شدة لدى الرجال، حيث تُعوّض وظيفة الجين الطافر لدى النساء بأليل سليم من الكروموسوم الثاني غير التالف.
الأساس الجيني لتطور التهاب الكلية الوراثي هو طفرات في جينات سلاسل ألفا من الكولاجين من النوع الرابع. هناك ست سلاسل ألفا معروفة من الكولاجين من النوع الرابع G: تقع جينات السلسلتين a5 وa6 (Col4A5 وCol4A5) على الذراع الطويلة للكروموسوم X في المنطقة 21-22q؛ وتقع جينات السلسلتين a3 وa4 (Col4A3 وCol4A4) على الكروموسوم الثاني؛ وتقع جينات السلسلتين a1 وa2 (Col4A1 وCol4A2) على الكروموسوم الثالث عشر.
في معظم الحالات (80-85%)، يُكتشف نمط وراثي مرتبط بالكروموسوم X للمرض، ويرتبط بتلف جين Col4A5 نتيجةً للحذف أو الطفرات النقطية أو اضطرابات الربط. حاليًا، تم اكتشاف أكثر من 200 طفرة في جين Col4A5، مسؤولة عن خلل في تخليق سلاسل a5 من الكولاجين من النوع الرابع. في هذا النوع من الوراثة، يظهر المرض لدى الأطفال من كلا الجنسين، ولكنه أكثر حدة لدى الأولاد.
الطفرات في مواقع جيني Col4A3 وCol4A4، المسؤولتين عن تخليق سلسلتي الكولاجين من النوع الرابع، هي طفرات جسمية موروثة. ووفقًا للدراسات، يُلاحظ النمط السائد في 16% من حالات التهاب الكلية الوراثي، بينما يُلاحظ النمط المتنحي في 6% من المرضى. يُعرف حوالي 10 متغيرات من طفرات جيني Col4A3 وCol4A4.
تؤدي الطفرات إلى خلل في عمليات تجميع الكولاجين من النوع الرابع، مما يؤدي إلى خلل في بنيته. يُعد الكولاجين من النوع الرابع أحد المكونات الرئيسية للغشاء القاعدي الكبيبي، وجهاز القوقعة، وعدسة العين، وسيتم الكشف عن أمراضه في عيادة التهاب الكلية الوراثي.
يتكون الكولاجين من النوع الرابع، وهو جزء من الغشاء القاعدي الكبيبي، بشكل رئيسي من سلسلتين a1 (IV) وسلسلة a2 (IV)، ويحتوي أيضًا على سلاسل a3 وa4 وa5. في أغلب الأحيان، في الوراثة المرتبطة بالكروموسوم X، يصاحب طفرة جين Col4A5 غياب سلاسل a3 وa4 وa5 وa6 في بنية الكولاجين من النوع الرابع، ويزداد عدد سلسلتي o1 وa2 في الغشاء القاعدي الكبيبي. آلية هذه الظاهرة غير واضحة، ويُفترض أن سببها هو تغيرات ما بعد النسخ في mRNA.
يؤدي غياب سلاسل a3 وa4 وa5 في بنية الكولاجين من النوع الرابع في الأغشية القاعدية الكبيبية إلى ترققها وهشاشتها في المراحل المبكرة من متلازمة ألبورت، والتي تتجلى سريريًا بشكل أكثر شيوعًا في وجود بيلة دموية (وأقل شيوعًا في وجود بيلة دموية مصحوبة ببيلة بروتينية أو بروتينية فقط)، وفقدان السمع، وظهور عدسية مخروطية. يؤدي تفاقم المرض إلى زيادة سماكة الأغشية القاعدية وضعف نفاذيتها في المراحل المتأخرة، مع تكاثر نوعي الكولاجين الخامس والسادس فيها، مما يتجلى في زيادة وجود بيلة بروتينية وانخفاض وظائف الكلى.
تُحدد طبيعة الطفرة الكامنة وراء التهاب الكلية الوراثي إلى حد كبير مظهره الظاهري. في حالة حذف الكروموسوم X مع طفرة متزامنة في جيني Col4A5 وCol4A6 المسؤولين عن تخليق سلسلتي الكولاجين من النوع الرابع، تصاحب متلازمة ألبورت ورم عضلي ملساء في المريء والأعضاء التناسلية. ووفقًا لبيانات الأبحاث، في حالة حدوث طفرة في جين Col4A5 مرتبطة بحذف، تُلاحظ شدة أكبر للعملية المرضية، وهي مزيج من تلف كلوي مع أعراض خارج كلوية وتطور مبكر للفشل الكلوي المزمن، مقارنةً بطفرة نقطية في هذا الجين.
من الناحية الشكلية، يكشف المجهر الإلكتروني عن ترقق وطبقية الأغشية القاعدية الكبيبية (وخاصةً الصفيحة الكثيفة) ووجود حبيبات كثيفة الإلكترونات. قد تكون الآفات الكبيبية غير متجانسة لدى المريض نفسه، بدءًا من آفات مسراقية بؤرية ضئيلة وصولًا إلى تصلب الكبيبات. يكون التهاب الكبيبات في متلازمة ألبورت دائمًا سلبيًا للمناعة، مما يميزه عن التهاب كبيبات الكلى. تشمل السمات المميزة تطور ضمور أنبوبي، وتسلل لمفاوي، ووجود "خلايا رغوية" مع شوائب دهنية - عاثيات الدهون. مع تقدم المرض، يُكشف عن سماكة وتدمير واضح للأغشية القاعدية الكبيبية.
تظهر بعض التغيرات في الجهاز المناعي. يعاني مرضى التهاب الكلية الوراثي من انخفاض مستوى IgA، وميل لزيادة تركيز IgM في الدم. قد يرتفع مستوى IgG في المراحل المبكرة من المرض، وينخفض في المراحل المتأخرة. ربما تكون زيادة تركيز IgM وG بمثابة رد فعل تعويضي لنقص IgA.
يتم تقليل النشاط الوظيفي لنظام الخلايا الليمفاوية التائية؛ ويلاحظ انخفاض انتقائي في الخلايا الليمفاوية البائية المسؤولة عن تخليق Ig A، ويتم تعطيل الارتباط البلعمي للمناعة، ويرجع ذلك أساسًا إلى تعطيل عمليات التاكسي الكيميائي والهضم داخل الخلايا في الخلايا المتعادلة.
عند فحص خزعة الكلى لدى مرضى متلازمة ألبورت، تكشف بيانات المجهر الإلكتروني عن تغيرات هيكلية دقيقة في الغشاء القاعدي الكبيبي: ترقق، وتمزق في بنيته، وانقسام في الأغشية القاعدية الكبيبية مع تغير في سمكها وتفاوت شكلها. في المراحل المبكرة من التهاب الكلية الوراثي، يُحدد هذا العيب ترقق وهشاشة الأغشية القاعدية الكبيبية.
يُعد ترقق الأغشية الكبيبية علامةً أكثر وضوحًا، وهو أكثر شيوعًا لدى الفتيات. أما العلامة الأكثر ثباتًا في الفحص المجهري الإلكتروني لالتهاب الكلية الوراثي فهي انقسام الغشاء القاعدي، وترتبط شدة تلفه بشدة العملية.
أعراض متلازمة ألبورت عند الأطفال
غالبًا ما تُكتشف الأعراض الأولى لمتلازمة ألبرت، على شكل متلازمة بولية معزولة، لدى الأطفال في السنوات الثلاث الأولى من العمر. في معظم الحالات، يُكتشف المرض بالصدفة. تُكتشف متلازمة البول أثناء الفحص الوقائي للطفل، قبل دخوله دار رعاية الأطفال، أو أثناء الإصابة بالتهابات الجهاز التنفسي الحادة (ARVI). في حالة وجود خلل في البول أثناء الإصابة بالتهابات الجهاز التنفسي الحادة (ARVI). في التهاب الكلية الوراثي، على عكس التهاب كبيبات الكلى المكتسب، لا توجد فترة كامنة.
في المرحلة الأولى من المرض، لا تتأثر صحة الطفل بشكل كبير، ومن السمات المميزة استمرار متلازمة البول ومقاومتها. من أهم أعراضها وجود بيلة دموية بدرجات متفاوتة من الشدة، وهي تُلاحظ في 100% من الحالات. تُلاحظ زيادة في درجة البيلة الدموية أثناء أو بعد التهابات الجهاز التنفسي، أو النشاط البدني، أو بعد التطعيمات الوقائية. في معظم الحالات، لا تتجاوز نسبة البروتين في البول غرامًا واحدًا يوميًا، وقد تكون في بداية المرض غير منتظمة، ومع تقدم المرض، تزداد نسبة البروتين في البول. قد تظهر بيلة كريات الدم البيضاء، مع غلبة الخلايا الليمفاوية، بشكل دوري في الرواسب البولية، وهو ما يرتبط بتطور التغيرات الخلالية.
بعد ذلك، تضعف وظائف الكلى جزئيًا، وتتدهور الحالة العامة للمريض: التسمم، وضعف العضلات، وانخفاض ضغط الدم الشرياني، وغالبًا ضعف السمع (خاصة عند الأولاد)، وأحيانًا ضعف البصر. يتجلى التسمم بالشحوب والتعب والصداع. في المرحلة الأولية من المرض، لا يُكتشف فقدان السمع في معظم الحالات إلا من خلال تخطيط السمع. يمكن أن يحدث فقدان السمع في متلازمة ألبورت في فترات مختلفة من الطفولة، ولكن غالبًا ما يتم تشخيص فقدان السمع في سن 6-10 سنوات. يبدأ فقدان السمع عند الأطفال بترددات عالية، ويصل إلى درجة كبيرة في التوصيل الهوائي والعظمي، وينتقل من فقدان السمع الموصل للصوت إلى فقدان السمع المُستشعر للصوت. يمكن أن يكون فقدان السمع أحد الأعراض الأولى للمرض ويمكن أن يسبق متلازمة المسالك البولية.
في 20% من الحالات، يعاني مرضى متلازمة ألبورت من تغيرات في الأعضاء البصرية. أكثر التشوهات التي يتم اكتشافها شيوعًا هي تلك الخاصة بالعدسة: كروية العين، والمخروطية العدسية الأمامية أو الخلفية أو المختلطة، وإعتام عدسة العين المتنوع. في العائلات المصابة بمتلازمة ألبورت، هناك تواتر كبير لقصر النظر. يلاحظ عدد من الباحثين باستمرار تغيرات ثنائية حول العين في هذه العائلات على شكل حبيبات بيضاء أو صفراء زاهية في الجسم الأصفر. ويعتبرون هذه العلامة من الأعراض الثابتة ذات القيمة التشخيصية العالية في متلازمة ألبورت. وجد KS Chugh وآخرون (1993) في دراسة طب العيون لدى مرضى متلازمة ألبورت انخفاضًا في حدة البصر في 66.7% من الحالات، والمخروطية العدسية الأمامية في 37.8%، والبقع الشبكية في 22.2%، وإعتام عدسة العين في 20%، والقرنية المخروطية في 6.7%.
لدى بعض الأطفال المصابين بالتهاب الكلية الوراثي، وخاصةً عند الإصابة بالفشل الكلوي، يُلاحظ تأخر ملحوظ في النمو البدني. ومع تقدم الفشل الكلوي، يُصابون بارتفاع ضغط الدم الشرياني. ويُكتشف هذا المرض لدى الأطفال غالبًا في مرحلة المراهقة والفئات العمرية الأكبر.
يتميز مرضى التهاب الكلية الوراثي بوجود وصمات مختلفة (أكثر من 5-7) لخلل تكوّن النسيج الضام. ومن بين وصمات النسيج الضام لدى المرضى، يُعدّ فرط تباعد العينين، وارتفاع سقف الحنك، وتشوهات العض، وشكل الأذنين غير الطبيعي، وانحناء الخنصر في اليدين، و"فجوة الصندل" في القدمين أكثر شيوعًا. يتميز التهاب الكلية الوراثي بتماثل وصمات خلل تكوّن النسيج الضام داخل العائلة، بالإضافة إلى ارتفاع تواتر انتشارها بين أقارب المرضى الذين ينتقل المرض إليهم.
في المراحل المبكرة من المرض، يُكتشف انخفاض معزول في وظائف الكلى الجزئية: نقل الأحماض الأمينية، والكهارل، ووظيفة التركيز، وتكوّن الأحماض. تؤثر التغيرات اللاحقة على الحالة الوظيفية لكل من الأجزاء القريبة والبعيدة من النيفرون، وتتميز باضطرابات جزئية مشتركة. يحدث انخفاض في الترشيح الكبيبي لاحقًا، وغالبًا في مرحلة المراهقة. مع تطور التهاب الكلية الوراثي، يتطور فقر الدم.
وهكذا، يتميز التهاب الكلية الوراثي بمسارٍ مُتدرّج للمرض: بدايةً، مرحلة كامنة أو أعراض سريرية خفية، تتجلى بتغيرات طفيفة في متلازمة المسالك البولية، ثم يحدث تراجع تدريجي للعملية مع انخفاض في وظائف الكلى مصحوبًا بأعراض سريرية واضحة (التسمم، والوهن، وتأخر النمو، وفقر الدم). عادةً ما تظهر الأعراض السريرية بغض النظر عن تدرّج التفاعل الالتهابي.
يمكن أن يظهر التهاب الكلية الوراثي في فترات عمرية مختلفة، وهذا يعتمد على عمل الجين، الذي يكون في حالة مكبوتة حتى وقت معين.
تصنيف
هناك ثلاثة أنواع من التهاب الكلية الوراثي
- الخيار الأول - يتجلى سريريًا بالتهاب كلوي مصحوب ببول دموي، وفقدان سمع، وتلف في العين. يتطور التهاب الكلية تدريجيًا مع تطور الفشل الكلوي المزمن. نوع الوراثة سائد، ويرتبط بالكروموسوم X. من الناحية الشكلية، يُكشف عن خلل في بنية الغشاء القاعدي، وترققه، وانقسامه.
- الخيار الثاني - يتجلى سريريًا بالتهاب كلوي مصحوب ببيلة دموية دون فقدان السمع. يتطور التهاب الكلية تدريجيًا مع تطور الفشل الكلوي المزمن. نوع الوراثة سائد، ويرتبط بالكروموسوم X. من الناحية الشكلية، يُكتشف ترقق الغشاء القاعدي الشعري الكبيبي (وخاصةً الغشاء الصفيحي).
- الخيار الثالث - بيلة دموية عائلية حميدة. مسار المرض طبيعي، ولا يُصاب المريض بفشل كلوي مزمن. نوع الوراثة إما جسمي سائد أو جسمي متنحي. في حالة الوراثة الجسمية المتنحية، يُلاحظ مسار أشد للمرض لدى النساء.
تشخيص متلازمة ألبورت
وقد تم اقتراح المعايير التالية:
- وجود مريضين على الأقل مصابين باعتلال الكلية في كل عائلة؛
- البول الدموي هو العرض الرئيسي لاعتلال الكلية لدى المريض؛
- وجود فقدان السمع لدى أحد أفراد العائلة على الأقل؛
- تطور الفشل الكلوي المزمن لدى أحد الأقارب أو أكثر.
في تشخيص مختلف الأمراض الوراثية والخلقية، يُعطى نهج شامل للفحص، والأهم من ذلك، الاهتمام بالبيانات المُحصّلة عند تجميع شجرة نسب الطفل، أهمية كبيرة. يُعتبر تشخيص متلازمة ألبورت صحيحًا في الحالات التي تُكتشف فيها ثلاث من أربع علامات نموذجية لدى المريض: وجود بيلة دموية وفشل كلوي مزمن في العائلة، ووجود فقدان سمع عصبي حسي، واضطرابات بصرية لدى المريض، واكتشاف علامات انشقاق الغشاء القاعدي الكبيبي مع تغير في سمكه وتضاريسه غير المتساوية، وذلك من خلال خصائص المجهر الإلكتروني للخزعة.
يجب أن يشمل فحص المريض أساليب البحث السريرية والوراثية؛ ودراسة مستهدفة للتاريخ المرضي؛ والفحص العام للمريض مع مراعاة المعايير التشخيصية المهمة. في مرحلة التعويض، لا يمكن الكشف عن الأمراض إلا من خلال التركيز على متلازمات مثل وجود عبء وراثي، وانخفاض ضغط الدم، ووصمات متعددة لخلل تكوين الأجنة، والتغيرات في متلازمة المسالك البولية. في مرحلة عدم التعويض، قد تظهر أعراض خارج الكلية، مثل التسمم الحاد، والوهن، وتأخر النمو البدني، وفقر الدم، والتي تتجلى وتكثف مع انخفاض تدريجي في وظائف الكلى. في معظم المرضى، مع انخفاض وظائف الكلى، يُلاحظ ما يلي: انخفاض تكوين الأحماض والأمين؛ يلاحظ 50٪ من المرضى انخفاضًا كبيرًا في الوظيفة الإفرازية للكلى؛ نطاق محدود من التقلبات في الكثافة البصرية للبول؛ اضطراب في إيقاع الترشيح، ثم انخفاض في الترشيح الكبيبي. يتم تشخيص مرحلة الفشل الكلوي المزمن عندما يكون لدى المرضى مستوى مرتفع من اليوريا في مصل الدم (أكثر من 0.35 جم / لتر) لمدة 3-6 أشهر أو أكثر، وانخفاض في الترشيح الكبيبي إلى 25٪ من المعدل الطبيعي.
ينبغي إجراء التشخيص التفريقي لالتهاب الكلية الوراثي بشكل أساسي مع التهاب كبيبات الكلى المكتسب المصحوب ببيلة دموية. غالبًا ما يبدأ التهاب كبيبات الكلى المكتسب بشكل حاد، بعد فترة تتراوح بين أسبوعين وثلاثة أسابيع من الإصابة، مع ظهور أعراض خارج الكلية، بما في ذلك ارتفاع ضغط الدم منذ الأيام الأولى (على العكس، في التهاب الكلية الوراثي، انخفاض ضغط الدم)، وانخفاض الترشيح الكبيبي عند بداية المرض، وعدم وجود أي خلل في وظائف الأنابيب الكلوية الجزئية، بينما تظهر هذه الأعراض في التهاب الكلية الوراثي. يصاحب التهاب كبيبات الكلى المكتسب بيلة دموية وبروتينية أكثر وضوحًا، مع زيادة في معدل ترسيب كرات الدم الحمراء. تُعد التغيرات النموذجية في الغشاء القاعدي الكبيبي، المميزة لالتهاب الكلية الوراثي، ذات قيمة تشخيصية.
يُجرى التشخيص التفريقي لاعتلال الكلية الأيضي مع الفشل الكلوي المزمن، وفي حال وجود أمراض كلوية غير متجانسة سريريًا في العائلة، قد يكون هناك طيف من اعتلال الكلية يتراوح من التهاب الحويضة والكلية إلى حصوات المسالك البولية. غالبًا ما يشتكي الأطفال من ألم في البطن، وأحيانًا أثناء التبول، ورواسب في البول - أكسالات.
في حالة الاشتباه في الإصابة بالتهاب الكلية الوراثي، يجب تحويل المريض إلى قسم متخصص في أمراض الكلى لتوضيح التشخيص.
ما الذي يجب فحصه؟
ما هي الاختبارات المطلوبة؟
من الاتصال؟
علاج متلازمة ألبورت
يتضمن النظام الغذائي قيودًا على بذل مجهود بدني شاق والتعرض للهواء النقي. يجب أن يكون النظام الغذائي متكاملًا، ويحتوي على كميات كافية من البروتينات والدهون والكربوهيدرات الكاملة، مع مراعاة وظائف الكلى. ومن الأهمية بمكان اكتشاف بؤر العدوى المزمنة وعلاجها. تُستخدم الأدوية التالية: ATP، كوكاربوكسيلاز، بيريدوكسين (حتى 50 ملغ/يوم)، وكلوريد الكارنيتين. تُعطى الدورات العلاجية مرتين إلى ثلاث مرات سنويًا. أما في حالة البول الدموي، فيُوصف العلاج بالأعشاب - نبات القراص، وعصير التوت البري، واليارو.
هناك تقارير في الأدبيات الأجنبية والمحلية حول العلاج بالبريدنيزولون واستخدام مُثبِّطات الخلايا. ومع ذلك، يصعب تقييم تأثيره.
في حالة الفشل الكلوي المزمن يتم اللجوء إلى غسيل الكلى وزراعة الكلى.
لا توجد طرق علاجية محددة (فعّالة ممرضًا) لالتهاب الكلية الوراثي. تهدف جميع التدابير العلاجية إلى الوقاية من تدهور وظائف الكلى وإبطاءه.
يجب أن يكون النظام الغذائي متوازنًا وغنيًا بالسعرات الحرارية، مع مراعاة الحالة الوظيفية للكلى. في حال عدم وجود اضطرابات وظيفية، يجب أن يحتوي نظام الطفل الغذائي على كميات كافية من البروتينات والدهون والكربوهيدرات. في حال ظهور علامات خلل في وظائف الكلى، يجب تقليل كمية البروتين والكربوهيدرات والكالسيوم والفوسفور، مما يُؤخر تطور الفشل الكلوي المزمن.
ينبغي الحد من النشاط البدني، ويُنصح الأطفال بتجنب ممارسة الرياضة.
ينبغي تجنب مخالطة المرضى المصابين بالعدوى، وتقليل خطر الإصابة بأمراض الجهاز التنفسي الحادة. كما أن تعقيم بؤر العدوى المزمنة ضروري. لا تُعطى التطعيمات الوقائية للأطفال المصابين بالتهاب الكلية الوراثي، ويُتاح التطعيم فقط في الحالات الوبائية.
العلاج الهرموني ومثبطات المناعة غير فعال في التهاب الكلية الوراثي. هناك مؤشرات على تأثير إيجابي (انخفاض في بروتينية البول وإبطاء تطور المرض) مع الاستخدام طويل الأمد لمثبطات السيكلوسبورين أ ومثبطات الإنزيم المحول للأنجيوتنسين (ACE).
في علاج المرضى يتم استخدام الأدوية التي تعمل على تحسين عملية التمثيل الغذائي:
- البيريدوكسين - 2-3 ملغ/كغ/يوم في 3 جرعات لمدة 4 أسابيع؛
- - كوكاربوكسيليز - 50 ملغ عضليًا كل يومين، بإجمالي 10-15 حقنة؛
- ATP - 1 مل في العضل كل يومين، 10-15 حقنة؛
- فيتامين أ - 1000 وحدة دولية/سنة/يوم في جرعة واحدة لمدة أسبوعين؛
- فيتامين E - 1 ملغ/كغ/يوم في جرعة واحدة لمدة أسبوعين.
يساعد هذا النوع من العلاج على تحسين الحالة العامة للمرضى وتقليل الخلل في الأنابيب ويتم إجراؤه على شكل دورات 3 مرات في السنة.
يمكن استخدام الليفاميزول كمنظم للمناعة - 2 ملغ/كغ/يوم 2-3 مرات في الأسبوع مع فترات راحة بين الجرعات لمدة 3-4 أيام.
وفقًا لبيانات الأبحاث، فإن الأكسجين عالي الضغط له تأثير إيجابي على شدة البول الدموي واختلال وظائف الكلى.
الطريقة الأكثر فعالية لعلاج التهاب الكلية الوراثي هي زراعة الكلى في الوقت المناسب. في هذه الحالة، لا تحدث انتكاسة للمرض بعد عملية الزرع؛ وفي نسبة ضئيلة (حوالي 5%)، قد يحدث التهاب كلية في الكلية المزروعة مرتبطًا بمستضدات الغشاء القاعدي الكبيبي.
يُعدّ التشخيص قبل الولادة والعلاج بالهندسة الوراثية اتجاهًا واعدًا. تُظهر التجارب على الحيوانات كفاءة عالية في نقل الجينات السليمة المسؤولة عن تخليق سلاسل ألفا الكولاجينية من النوع الرابع إلى أنسجة الكلى، وبعد ذلك يُلاحَظ تخليق هياكل الكولاجين الطبيعية.
تنبؤ بالمناخ
إن تشخيص التهاب الكلية الوراثي يكون دائمًا خطيرًا.
المعايير غير المواتية من الناحية التشخيصية لمسار التهاب الكلية الوراثي هي:
- الجنس الذكر؛
- التطور المبكر لفشل الكلى المزمن لدى أفراد العائلة؛
- بروتينية في البول (أكثر من 1 جرام/يوم)؛
- سماكة الأغشية القاعدية الكبيبية حسب المجهر؛
- التهاب العصب السمعي؛
- الحذف في جين Col4A5.
إن تشخيص البول الدموي العائلي الحميد هو أكثر إيجابية.
Использованная литература