
مرض شاركو ماري توث
آخر مراجعة: 23.11.2021

تتم مراجعة جميع محتويات iLive طبياً أو التحقق من حقيقة الأمر لضمان أكبر قدر ممكن من الدقة الواقعية.
لدينا إرشادات صارمة من مصادرنا ونربط فقط بمواقع الوسائط ذات السمعة الطيبة ، ومؤسسات البحوث الأكاديمية ، وطبياً ، كلما أمكن ذلك استعراض الأقران الدراسات. لاحظ أن الأرقام الموجودة بين قوسين ([1] و [2] وما إلى ذلك) هي روابط قابلة للنقر على هذه الدراسات.
إذا كنت تشعر أن أيًا من المحتوى لدينا غير دقيق أو قديم. خلاف ذلك مشكوك فيه ، يرجى تحديده واضغط على Ctrl + Enter.
ضمور العضلات الشظية أو متلازمة أو مرض شاركو ماري توث هو مجموعة كاملة من الأمراض الوراثية المزمنة التي تسبب تلفًا في الأعصاب الطرفية.
وفقًا لـ ICD-10 في قسم أمراض الجهاز العصبي ، فإن رمز هذا المرض هو G60.0 (اعتلال الأعصاب الوراثي الحركي والحسي). كما أنه مدرج في قائمة الأمراض اليتيمة.
علم الأوبئة
وفقًا للإحصاءات السريرية ، فإن معدل انتشار جميع أنواع مرض شاركو ماري توث لكل 100 ألف من السكان هو 19 حالة (وفقًا لمصادر أخرى ، حالة واحدة لكل 2.5-10 آلاف نسمة).
يمثل النوع 1 من CMT حوالي ثلثي الحالات (حالة واحدة لكل 5-7 آلاف من السكان) ، وتقريبًا 70٪ منهم مرتبط بتكرار الجين PMP22. في العالم ، يصيب هذا النوع من الأمراض أكثر من 1.2 مليون شخص.
تقدر نسبة حدوث النوع 4 CMT بحوالي 1-5 حالات لكل 10 آلاف طفل. [1]
الأسباب مرض شاركو ماري توث
وفقًا لتصنيف متلازمات الأعصاب المتعددة ، فإن ضمور العضلات الشظوي (الشظوي) أو ضمور العضلات العصبي شاركو ماري توث أو مرض شاركو ماري توث (اختصارًا باسم CMT) يشير إلى اعتلالات الأعصاب الحركية الحركية المحددة وراثيًا. [2]
أي أن أسباب حدوثها هي الطفرات الجينية. واعتمادًا على طبيعة التشوهات الجينية ، تختلف الأنواع أو الأنواع الرئيسية لهذه المتلازمة: إزالة الميالين والمحاور. تشمل المجموعة الأولى مرض شاركو ماري توث من النوع الأول (CMT1) ، والذي يحدث نتيجة ازدواج جين PMP22 على الكروموسوم 17 ، والذي يشفر بروتين المايلين المحيطي عبر الغشاء 22. نتيجة لذلك ، إزالة الميالين القطعي للغلاف المحوري (عمليات الخلايا العصبية) وانخفاض في سرعة التوصيل العصبي. تحدث الإشارات. بالإضافة إلى ذلك ، قد تكون هناك طفرات في بعض الجينات الأخرى.
الشكل المحوري هو مرض شاركو ماري توث من النوع 2 (CMT2) ، والذي يؤثر على المحاور نفسها ويرتبط بالتغيرات المرضية في جين MFN2 في موضع 1p36.22 ، والذي يشفر بروتين الغشاء ميتوفوسين -2 ، وهو أمر ضروري لدمج الميتوكوندريا وتشكيل شبكات الميتوكوندريا الوظيفية داخل الخلايا العصبية المحيطية. هناك أكثر من عشرة أنواع فرعية من CMT2 (مع حدوث طفرات في جينات معينة).
وتجدر الإشارة إلى أنه قد تم تحديد أكثر من مائة جين حتى الآن ، تسبب أضرارها ، الموروثة ، في أنواع فرعية مختلفة من مرض شاركو ماري توث. على سبيل المثال ، الطفرات في جين RAB7 تطور النوع 2B CMT ؛ يؤدي تغيير جين SH3TC2 (الذي يشفر أحد بروتينات أغشية خلايا شوان) إلى النوع 4C CMT ، والذي يتجلى في مرحلة الطفولة ويتميز بإزالة الميالين من الخلايا العصبية الحركية والحسية (دزينة ونصف من أشكال النوع 4 من هذا المرض مميز).
نوع نادر من SMT 3 (يسمى متلازمة Dejerine-Sott) ، الناجم عن طفرات في جينات PMP22 و MPZ و EGR2 وغيرها ، يبدأ أيضًا في التطور في مرحلة الطفولة المبكرة.
عندما يحدث نوع CMT 5 في سن 5-12 سنة ، لا يلاحظ فقط اعتلال الأعصاب الحركية (في شكل شلل سفلي تشنجي في الأطراف السفلية) ، ولكن أيضًا تلف الأعصاب البصرية والسمعية.
ضعف العضلات وضمور العصب البصري (مع فقدان البصر) ، وكذلك مشاكل التوازن ، من سمات النوع 6 من CMT. ومع مرض شاركو ماري توث من النوع السابع ، لا يُلاحَظ فقط اعتلال الأعصاب الحركية الحركية ، بل يُلاحظ أيضًا مرض الشبكية في شكل التهاب الشبكية الصباغي.
يعتبر مرض SMT أو مرض شاركو ماري-توث الأكثر شيوعًا والمرتبط برباعي الأطراف (ضعف حركة الذراعين والساقين) بين الرجال من النوع المزيل للميالين ويعتبر نتيجة لطفرة في جين GJB1 على الذراع الطويلة للكروموسوم X ، الذي يرمز إلى connexin 32 ، وهو بروتين عبر الغشاء خلايا Schwann وخلايا oligodendrocytes ، والذي ينظم نقل الإشارات العصبية. [3]
عوامل الخطر
عامل الخطر الرئيسي لـ CMT هو وجود هذا المرض في تاريخ عائلي ، أي في الأقارب.
وفقًا لعلماء الوراثة ، إذا كان كلا الوالدين حاملين للجين المتنحي الجسدي لمرض شاركو ماري توث ، فإن خطر إنجاب طفل سيصاب بهذا المرض هو 25٪. وتقدر خطورة أن يحمل الطفل هذا الجين (ولكن لن تظهر عليه أي أعراض) بنسبة 50٪.
في حالة الوراثة المرتبطة بـ X (عندما يكون الجين المتحور على كروموسوم X الخاص بالمرأة) ، هناك خطر بنسبة 50٪ أن الأم سوف تنقل هذا الجين إلى ابنها ، وسوف يصاب بمرض CMT. عند ولادة طفلة ، قد لا يحدث المرض ، لكن يمكن لأبناء البنت (الأحفاد) أن يرثوا الجين المعيب - مع تطور المرض.
طريقة تطور المرض
في أي نوع من مرض شاركو ماري توث ، ترجع أسبابه إلى شذوذ وراثي في الأعصاب المحيطية: المحرك (المحرك) والحسي (الحسي).
إذا كان نوع CMT مزيل للميالين ، فإن تدمير أو عيب غمد الميالين ، الذي يحمي محاور الأعصاب الطرفية ، يؤدي إلى تباطؤ في انتقال النبضات العصبية للجهاز العصبي المحيطي - بين الدماغ والعضلات والأعضاء الحسية.
في النوع المحوري للمرض ، تتأثر المحاور بشكل مباشر ، مما يؤثر سلبًا على قوة الإشارات العصبية ، وهو ما لا يكفي للتحفيز الكامل للعضلات والأعضاء الحسية.
اقرأ أيضًا:
كيف تنتشر متلازمة شاركو ماري توث؟ يمكن توريث الجينات المعيبة بطريقة جسمية سائدة أو صفة متنحية.
الأكثر شيوعًا - الوراثة الصبغية السائدة - يحدث عندما يكون هناك نسخة واحدة من الجين المتحور (يحمله أحد الوالدين). ويقدر احتمال انتقال CMT لكل من الأطفال المولودين بنسبة 50٪. [4]
في الوراثة الجسدية المتنحية ، يتطلب المرض نسختين من الجين المعيب (واحدة من كل والد ليس لديه علامات المرض).
في 40-50٪ من الحالات ، يحدث إزالة الميالين الوراثي السائد ، أي CMT النوع 1 ؛ في 12-26٪ من الحالات - CMT المحوري ، أي النوع 2. وفي 10-15٪ من الحالات ، يتم ملاحظة الوراثة المرتبطة بـ X. [5]
الأعراض مرض شاركو ماري توث
عادة ، تبدأ العلامات الأولى لهذا المرض في الظهور في مرحلة الطفولة والمراهقين وتتطور تدريجياً طوال الحياة ، على الرغم من أن المتلازمة يمكن أن تظهر في وقت لاحق. إن مجموعة الأعراض متغيرة ، ولا يمكن التنبؤ بمعدل تطور المرض وشدته.
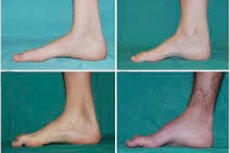
هناك أعراض نموذجية في المرحلة الأولية مثل زيادة التعب العام. انخفاض توتر (ضعف) عضلات القدمين والكاحلين وأسفل الساقين ؛ قلة ردود الفعل. هذا يجعل من الصعب تحريك القدم ويؤدي إلى عسر التنسج (اضطراب المشي) في شكل ارتفاع أعلى للساقين ، غالبًا مع التعثر والسقوط المتكرر. قد تظهر علامات مرض شاركو-ماري-توث عند الطفل الصغير خراقة وصعوبة في المشي ، وهو أمر غير معتاد بالنسبة للعمر ، ويرتبط بقدم متدلية ثنائية الأطراف . تشوهات القدم مميزة أيضًا: قوس مرتفع (قدم مجوفة) أو أقدام مسطحة قوية ، أصابع منحنية (تشبه المطرقة).
في حالة المشي على أصابع القدم على خلفية انخفاض ضغط الدم العضلي ، قد يشك طبيب الأعصاب في إصابة الطفل بنوع 4 من CMT ، حيث قد لا يتمكن الأطفال في سن المراهقة من المشي.
مع تقدمه ، ينتشر ضمور العضلات وضعفها إلى الأطراف العلوية ، مما يجعل من الصعب على المهارات الحركية الدقيقة وأنشطة اليد العادية. يشير انخفاض الإحساس اللمسي والقدرة على الشعور بالدفء والبرودة ، بالإضافة إلى التنميل في القدمين واليدين ، إلى تلف محاور الأعصاب الحسية.
يتجلى في مرض شاركو ماري توث في مرحلة الطفولة من النوعين 3 و 6 ، وهناك ترنح حساس (ضعف تنسيق الحركات والتوازن) ، وارتعاش عضلي ورعاش ، وتلف في العصب الوجهي ، وضمور بصري مع رأرأة ، وفقدان السمع.
في مراحل لاحقة ، قد يكون هناك رعشات لا يمكن السيطرة عليها (رعشات) وتشنجات عضلية متكررة ؛ يمكن أن تؤدي مشاكل الحركة إلى تطور الألم: العضلات والمفاصل والاعتلال العصبي.
المضاعفات والنتائج
يمكن أن يكون لمرض شاركو ماري توث مضاعفات وعواقب مثل:
- المزيد من الالتواء والكسور.
- تقلصات مرتبطة بقصر العضلات والأوتار حول المفصل.
- الجنف (انحناء العمود الفقري) ؛
- مشاكل في التنفس - مع تلف الألياف العصبية التي تعصب عضلات الحجاب الحاجز:
- فقدان القدرة على التحرك بشكل مستقل.
التشخيص مرض شاركو ماري توث
يشمل التشخيص الفحص السريري والتاريخ (بما في ذلك تاريخ العائلة) والفحص العصبي والجهازي.
يتم إجراء الاختبارات للتحقق من مدى الحركة والحساسية وردود الفعل الوترية. التوصيل العصبي يمكن تقييمها من قبل التشخيص مفيدة - الكهربائي أو electroneuromyography . قد تكون هناك حاجة أيضًا إلى الموجات فوق الصوتية أو التصوير بالرنين المغناطيسي. [6]
التشخيصات الوراثية أو الحمض النووي لتحديد الطفرات الجينية الأكثر شيوعًا التي تسبب CMT في عينة الدم محدودة ، حيث لا تتوفر اختبارات الحمض النووي حاليًا لجميع أنواع CMT. لمزيد من التفاصيل انظر - البحث الجيني
في بعض الحالات ، يتم أخذ خزعة من العصب المحيطي (عادةً عضلة الساق).
تشخيص متباين
يتم إجراء التشخيص التفريقي مع اعتلالات الأعصاب الطرفية الأخرى ، مثل الحثل العضلي الدوشيني ، ومتلازمات الاعتلال النخاعي والوهن العضلي ، والاعتلال العصبي السكري ، مع داء المشعرات النخاعية في حالة التصلب الجانبي المتعدد والضموري ، ومتلازمة غيلان باريه ، وصدمة العصب الشظوي وضمور القرص (بما في ذلك ضمور العمود الفقري) ) ، الأضرار التي لحقت المخيخ أو المهاد ، وكذلك الآثار الجانبية للعلاج الكيميائي (عند العلاج بمضادات التجلط الخلوي مثل فينكريستين أو باكليتاكسيل). [7]
من الاتصال؟
علاج او معاملة مرض شاركو ماري توث
اليوم ، يتكون علاج هذا المرض الوراثي من تمارين العلاج الطبيعي (التي تهدف إلى تقوية وتمديد العضلات) ؛ العلاج المهني (الذي يساعد المرضى الذين يعانون من ضعف عضلات اليدين) ؛ باستخدام أجهزة تقويم العظام لتسهيل المشي. إذا لزم الأمر ، تناول المسكنات أو مضادات الاختلاج. [8]
في حالات القدم المسطحة الواضحة ، يمكن إجراء قطع العظم ، وفي حالة تشوه الكعب ، يشار إلى تصحيحها الجراحي - إيثاق المفصل. [9]
البحث جار على كل من المكون الجيني للمرض وطرق علاجه. استخدام الخلايا الجذعية وبعض الهرمونات والليسيثين وحمض الأسكوربيك لم تسفر عن نتائج إيجابية بعد.
ولكن بفضل أحدث الأبحاث ، في المستقبل القريب ، قد يظهر بحث جديد حقًا في علاج مرض شاركو ماري توث. لذلك ، منذ عام 2014 ، تعمل الشركة الفرنسية Pharnext على تطوير ، ومنذ منتصف عام 2019 ، تجارب سريرية للدواء PXT3003 لعلاج CMT من النوع 1 عند البالغين ، مما يؤدي إلى قمع التعبير المتزايد عن جين PMP22 ، وتحسين الميالين من الأعصاب الطرفية و إضعاف الأعراض العصبية العضلية.
يعمل المتخصصون في شركة Sarepta Therapeutics الطبية (الولايات المتحدة الأمريكية) على علاج جيني لمرض شاركو ماري توث من النوع الأول. سيستخدم هذا العلاج فيروسًا غديًا غير ضار (AAV) من جنس Dependovirus مع جينوم DNA أحادي الخيط ، والذي سينقل جين NTF3 إلى الجسم ، والذي يشفر بروتين neurotrophin-3 (NT-3) الضروري لـ عمل الخلايا العصبية شوان.
بحلول نهاية عام 2020 ، ستبدأ Helixmith التجارب السريرية للعلاج الجيني Engensis (VM202) الذي تم تطويره في كوريا الجنوبية لعلاج أعراض العضلات في النوع 1 من CMT. [10]
الوقاية
يمكن أن تكون الوقاية من CMT استشارة وراثية للآباء المستقبليين ، خاصة إذا كان أحد الزوجين مصابًا بهذا المرض في العائلة. ومع ذلك ، فقد تم تحديد حالات الطفرات الجينية de novo ، أي في غياب المرض في تاريخ العائلة.
أثناء الحمل ، يسمح لك أخذ عينة من خلايا المشيمة (من 10 إلى 13 أسبوعًا من الحمل) ، بالإضافة إلى تحليل السائل الأمنيوسي (من 15 إلى 18 أسبوعًا) ، بالتحقق من احتمالية الإصابة بمرض شاركو ماري توث في الجنين.
توقعات
بشكل عام ، يعتمد تشخيص أنواع مختلفة من مرض شاركو ماري توث على الشدة السريرية ، ولكن على أي حال ، يتقدم المرض ببطء. يعاني العديد من المرضى من إعاقات ، على الرغم من أن هذا لا يقلل من متوسط العمر المتوقع.