خبير طبي في المقال

منشورات جديدة
داء هنتنغتون
آخر مراجعة: 05.07.2025

تتم مراجعة جميع محتويات iLive طبياً أو التحقق من حقيقة الأمر لضمان أكبر قدر ممكن من الدقة الواقعية.
لدينا إرشادات صارمة من مصادرنا ونربط فقط بمواقع الوسائط ذات السمعة الطيبة ، ومؤسسات البحوث الأكاديمية ، وطبياً ، كلما أمكن ذلك استعراض الأقران الدراسات. لاحظ أن الأرقام الموجودة بين قوسين ([1] و [2] وما إلى ذلك) هي روابط قابلة للنقر على هذه الدراسات.
إذا كنت تشعر أن أيًا من المحتوى لدينا غير دقيق أو قديم. خلاف ذلك مشكوك فيه ، يرجى تحديده واضغط على Ctrl + Enter.
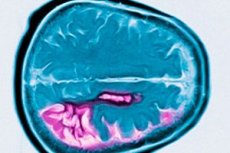
مرض هنتنغتون هو اضطراب عصبي تنكسي جسمي سائد، يتميز بتدهور إدراكي تدريجي، وحركات لا إرادية، وضعف في التنسيق الحركي، بدءًا من منتصف العمر. يُؤكد التشخيص بالفحوصات الجينية. يعتمد العلاج بشكل أساسي على الأعراض. قد يُوصى بإجراء الفحوصات الجينية لأقارب الدم. وصف جورج هنتنغتون هذه الحالة لأول مرة عام ١٨٧٢، بعد دراسة حالة عائلية لسكان لونغ آيلاند.
يبلغ معدل انتشار مرض هنتنغتون حوالي 10 حالات لكل 100000 نسمة، ونظرًا لظهوره المتأخر، فإن حوالي 30 شخصًا من أصل 100000 لديهم خطر بنسبة 50٪ للإصابة به في حياتهم. وعلى الرغم من أن المرض يظهر غالبًا بين سن 35 و40 عامًا، إلا أن النطاق العمري لبدء المرض واسع جدًا، حيث يكون أول ظهور في سن 3 سنوات وأحدث ظهور في سن 90 عامًا. وعلى الرغم من أنه كان يُعتقد في الأصل أن المرض يتمتع بنسبة اختراق 100٪، إلا أنه يُعتقد الآن أن هذا ليس هو الحال دائمًا. في الأفراد الذين ورثوا الجين للمرض من والدهم، يظهر المرض في المتوسط قبل 3 سنوات من أولئك الذين ورثوا الجين المرضي من والدتهم. وفي حوالي 80٪ من المرضى الذين ورثوا الجين المرضي من والدهم، يظهر المرض قبل سن 20. وتسمى ظاهرة الظهور المبكر لعيب وراثي في النسل بالتوقع.
 [ 1 ]
[ 1 ]
ما الذي يسبب مرض هنتنغتون؟
لا يُظهر مرض هنتنغتون أي تفضيل جنسي. يظهر ضمور النواة المذنبة، حيث تتحلل الخلايا العصبية الصغيرة وينخفض مستوى النواقل العصبية - حمض غاما أمينوبوتيريك (GABA) والمادة P.
جين متحور ذو عدد متزايد ("توسع") من تسلسلات الحمض النووي CAG (سيستين-ألانين-جلايسين) التي تُشفّر الحمض الأميني جلوتامين، هو المسؤول عن تطور داء هنتنغتون. يحتوي ناتج هذا الجين، وهو بروتين هنتنغتين كبير الحجم، على كمية زائدة من بقايا البولي جلوتامين، مما يؤدي إلى المرض بآلية غير معروفة. كلما زادت تكرارات CAG، ظهر المرض مبكرًا واشتدّ مساره. من جيل إلى جيل، يمكن أن يزداد عدد التكرارات، مما يؤدي مع مرور الوقت إلى تفاقم النمط الظاهري للعائلة.
على الرغم من الاهتمام الكبير بالتغيرات الجينية والكيميائية الحيوية في مرض باركنسون، إلا أن البحث عن جين مسؤول عن المرض لم يُكتب له النجاح حتى أواخر سبعينيات القرن الماضي. في ذلك الوقت، نظمت نانسي ويكسلر وآلان توبين ورشة عمل برعاية مؤسسة الأمراض الوراثية لمناقشة استراتيجية لإيجاد جين مسؤول عن مرض هنتنغتون. واقترح ديفيد هوسمان وديفيد بوتشتاين وراي وايت، الذين حضروا الاجتماع، أن تقنيات الحمض النووي المؤتلف المُطورة حديثًا قد تُساعد في تحقيق هذا الهدف. تمثلت إحدى المهام الرئيسية في المشروع في العثور على عائلة كبيرة مصابة بمرض هنتنغتون عبر أجيال عديدة للحصول على عينات من الحمض النووي. في عام ١٩٧٩، أُطلق مشروع مشترك بين علماء من فنزويلا والولايات المتحدة لفحص عائلة كبيرة مصابة بمرض هنتنغتون تعيش على شواطئ بحيرة ماراشيبو (فنزويلا). في عام ١٩٨٣، حُدد موقع جين داء هنتنغتون في نهاية الذراع القصير للكروموسوم ٤ (غوسيلا وآخرون، ١٩٨٣)، وبعد عقد من الزمان، كُشف أن طفرة هذا الجين تتمثل في زيادة عدد تكرارات ثلاثي نوكليوتيد السيتوزين-أدينين-جوانين (CAG) (مجموعة أبحاث داء هنتنغتون التعاونية، ١٩٩٣). تُعتبر المنهجية التي طورتها هذه المجموعة العلمية حاليًا معيارًا للاستنساخ الموضعي للجينات الجديدة.
في حين أن الجين البري يمتد من 10 إلى 28 تكرارًا لـ CAG، فإن الشكل المتحور للجين المسبب لمرض هنتنغتون يمتد بشكل متزايد من 39 إلى أكثر من 100 تكرار لـ CAG. ساعد اكتشاف توسع تكرارات ثلاثي النوكليوتيدات في تفسير العديد من السمات السريرية للمرض. وعلى وجه الخصوص، تم العثور على علاقة عكسية بين عمر البداية وطول المنطقة ذات النوكليوتيدات المتكررة. يمكن تفسير توقع الوراثة الأبوية بحقيقة أن زيادة في عدد التكرارات تحدث غالبًا لدى الرجال أثناء تكوين الحيوانات المنوية. أظهر تحليل الطفرات الجديدة أنها تحدث عادةً عندما يكون لدى أحد الوالدين، وعادةً الأب، عدد تكرارات CAG أعلى من 28؛ في هذه الحالة، زاد عدد هذه التكرارات في الجيل التالي. وقد ثبت الآن أنه إذا لم يتجاوز عدد التكرارات 28، فإنه ينتقل بشكل مستقر من جيل إلى جيل. إذا تراوح عدد التكرارات بين ٢٩ و٣٥، فلا تظهر أعراض داء هنتنغتون، ولكن عند انتقاله إلى الأبناء، قد يزداد طول هذه المنطقة. أما إذا تراوح عدد التكرارات بين ٣٦ و٣٩، فقد يظهر المرض سريريًا في بعض الحالات (وليس دائمًا) (اختراق غير كامل)، وعند انتقاله إلى الأبناء، قد تزداد عدد تكرارات ثلاثي النوكليوتيد. أما إذا تجاوز عدد التكرارات ٤٠، فإن المرض يظهر في معظم الحالات، وعند انتقاله إلى الأبناء، قد يزداد عدد التكرارات. ولا تزال أسباب زيادة عدد التكرارات مجهولة.
الشكل المرضي لمرض هنتنغتون
يتميز مرض هنتنغتون بفقدان الخلايا العصبية بشكل رئيسي في النواة المذنبة والبطامة، وإلى حد ما أيضًا في القشرة المخية وهياكل الدماغ الأخرى. ينخفض الوزن الإجمالي للدماغ في مرض هنتنغتون ليس فقط بسبب انخفاض عدد الخلايا العصبية، ولكن أيضًا بسبب فقدان المادة البيضاء. في القشرة المخية، تتأثر خلايا الطبقتين الخامسة والسادسة بشكل أكبر. ترتبط شدة التغيرات التنكسية المجهرية والعيانية (المعدلة حسب العمر عند الوفاة) بعدد تكرارات CAG. أظهر التحليل المرضي المفصل للتغيرات في مئات حالات مرض هنتنغتون أن تنكس الجسم المخطط يبدأ في الجزء الظهري الإنسي من النواة المذنبة والجزء الظهري الوحشي من البطامة، ثم ينتشر بطنيًا. تتأثر مجموعات مختلفة من الخلايا العصبية في النواة المذنبة والبطامة بدرجات متفاوتة. تبقى الخلايا العصبية البينية في الجسم المخطط سليمة نسبيًا، ولكن تتأثر بعض الخلايا العصبية الإسقاطية بشكل انتقائي. في الشكل الشبابي لمرض هنتنغتون، تكون التغيرات المرضية الشكلية في المخطط أكثر وضوحًا وانتشارًا، وتشمل القشرة المخية والمخيخ والمهاد والكرة الشاحبة.
التغيرات الكيميائية العصبية في مرض هنتنغتون
GABA. كشفت الدراسات الكيميائية العصبية لأدمغة مرضى داء هنتنغتون عن انخفاض ملحوظ في تركيز GABA في الجسم المخطط. وأكدت دراسات لاحقة أن داء هنتنغتون يرتبط بانخفاض في عدد الخلايا العصبية GABAergic، وأظهرت أن تركيزات GABA تنخفض ليس فقط في الجسم المخطط، ولكن أيضًا في مناطق إسقاطه - الأجزاء الخارجية والداخلية من الكرة الشاحبة والمادة السوداء. في دماغ مرضى هنتنغتون، تم الكشف أيضًا عن تغيرات في مستقبلات GABA باستخدام دراسات ربط المستقبلات والتهجين الموضعي للـ mRNA. انخفض عدد مستقبلات GABA بشكل معتدل في النواة المذنبة والبطامة، ولكنه زاد في الجزء الشبكي من المادة السوداء والجزء الخارجي من الكرة الشاحبة، والذي يُرجَّح أنه ناتج عن فرط الحساسية لفقدان التعصيب.
الأستيل كولين. يُستخدم الأستيل كولين كناقل عصبي بواسطة الخلايا العصبية الكبيرة غير الشوكية في الجسم المخطط. أظهرت الدراسات المبكرة بعد الوفاة على مرضى مصابين بداء هنتنغتون انخفاضًا في نشاط أسيتيل كولين ترانسفيراز (ChAT) في الجسم المخطط، مما يشير إلى فقدان الخلايا العصبية الكولينية. ومع ذلك، بالمقارنة مع الانخفاض الكبير في الخلايا العصبية GABAergic، فإن الخلايا العصبية الكولينية تبقى بمنأى نسبيًا. لذلك، فإن كثافة الخلايا العصبية الموجبة لأسيتيل كولين استريز ونشاط ChAT في الجسم المخطط مرتفعة نسبيًا مقارنةً بالمجموعة الضابطة المتطابقة في العمر.
المادة P. توجد المادة P في العديد من الخلايا العصبية الشوكية المتوسطة في الجسم المخطط، والتي تمتد بشكل رئيسي إلى الجزء الداخلي من الكرة الشاحبة والمادة السوداء، وعادةً ما تحتوي أيضًا على الدينورفين وحمض غاما أمينوبوتيريك (GABA). تنخفض مستويات المادة P في الجسم المخطط والجزء الشبكي من المادة السوداء في مرض هنتنغتون. في المرحلة النهائية من المرض، كشفت الدراسات المناعية الكيميائية عن انخفاض ملحوظ في عدد الخلايا العصبية التي تحتوي على المادة P. في المراحل المبكرة، تكون الخلايا العصبية التي تحتوي على المادة P والممتدة إلى الجزء الداخلي من الكرة الشاحبة أقل إصابةً نسبيًا، مقارنةً بالخلايا العصبية التي تمتد إلى الجزء الشبكي من المادة السوداء.
ببتيدات أفيونية. يوجد الإنكيفالين في الخلايا العصبية الغابايرجية متوسطة الشائكة ذات المسار غير المباشر، والتي تمتد إلى الجزء الخارجي من الكرة الشاحبة وتحمل مستقبلات D2. أظهرت الدراسات المناعية الكيميائية أن الخلايا العصبية المحتوية على الإنكيفالين والممتدة إلى الجزء الخارجي من الكرة الشاحبة تُفقد في مرحلة مبكرة من مرض هنتنغتون. ويبدو أن هذه الخلايا تموت أبكر من الخلايا المحتوية على المادة P والممتدة إلى الجزء الداخلي من الكرة الشاحبة.
الكاتيكولامينات. تقع الخلايا العصبية التي تحتوي على أمينات حيوية (الدوبامين، السيروتونين) والممتدة نحو الجسم المخطط في الجزء المُدمج من المادة السوداء، والغطاء البطني، ونواة الرافي. في حين أن النتوءات النورأدرينية نحو الجسم المخطط البشري ضئيلة، فإن مستويات السيروتونين والدوبامين (لكل غرام من الأنسجة) في الجسم المخطط مرتفعة، مما يشير إلى الحفاظ على هذه النتوءات الواردة على الرغم من الفقدان الملحوظ لخلايا الجسم المخطط. تبقى الخلايا العصبية الدوبامينية للمادة السوداء سليمة في كل من الشكلين الكلاسيكي والشبابي من داء هنتنغتون.
السوماتوستاتين/نيوروببتيد Y وسينثيتاز أكسيد النيتريك. أظهر قياس مستويات السوماتوستاتين والنيوروببتيد Y في الجسم المخطط في مرض هنتنغتون زيادةً قدرها 4-5 أضعاف مقارنةً بالأنسجة الطبيعية. أظهرت الدراسات المناعية الكيميائية الحفاظ التام على الخلايا العصبية الداخلية في الجسم المخطط التي تحتوي على نيوروببتيد Y والسوماتوستاتين وسينثيتاز أكسيد النيتريك. وبالتالي، فإن هذه الخلايا العصبية مقاومة للعملية المرضية.
الأحماض الأمينية المُثيرة. اقتُرح أن موت الخلايا الانتقائي في داء هنتنغتون يُعزى إلى تأثير عصبي سام يُحفزه الغلوتامات. تختلف مستويات الغلوتامات وحمض الكينولينيك (وهو سم عصبي داخلي المنشأ ناتج ثانوي عن استقلاب السيروتونين وناهض لمستقبلات الغلوتامات) في الجسم المخطط لداء هنتنغتون بشكل طفيف، إلا أن دراسة حديثة باستخدام مطيافية الرنين المغناطيسي كشفت عن زيادة في مستويات الغلوتامات في الجسم الحي. يزداد مستوى الإنزيم الدبقي المسؤول عن تخليق حمض الكينولينيك في الجسم المخطط لداء هنتنغتون بنحو 5 أضعاف مقارنةً بالمستوى الطبيعي، بينما يزداد نشاط الإنزيم الذي يضمن تحلل حمض الكينولينيك في داء هنتنغتون بنسبة 20-50% فقط. وبالتالي، قد يزداد تخليق حمض الكينولينيك في داء هنتنغتون.
أظهرت دراسات مستقبلات الأحماض الأمينية المُثيرة (EAA) في مرض هنتنغتون انخفاضًا ملحوظًا في عدد مستقبلات NMDA، وAMPA، والكينات، والغلوتامات الأيضية في الجسم المخطط، بالإضافة إلى مستقبلات AMPA والكينات في القشرة المخية. في المرحلة المتأخرة من مرض هنتنغتون، كانت مستقبلات NMDA غائبة تقريبًا، بينما لوحظ انخفاض ملحوظ في عدد هذه المستقبلات في المراحل ما قبل السريرية والمبكرة.
حساسية انتقائية. في مرض هنتنغتون، تُفقد أنواع معينة من خلايا الجسم المخطط بشكل انتقائي. تموت الخلايا العصبية الشوكية المتوسطة، التي تمتد إلى الجزء الخارجي من الكرة الشاحبة وتحتوي على حمض جاما أمينوبوتيريك (GABA) وإنكيفالين، في مرحلة مبكرة جدًا من المرض، وكذلك الخلايا العصبية التي تحتوي على حمض جاما أمينوبوتيريك (GABA) والمادة P وتمتد إلى الجزء الشبكي من المادة السوداء. يؤدي فقدان الخلايا العصبية التي تحتوي على حمض جاما أمينوبوتيريك (GABA) وإنكيفالين وتمتد إلى الجزء الخارجي من الكرة الشاحبة إلى تثبيط هذه البنية، مما يؤدي بدوره إلى تثبيط نشط للنواة تحت المهاد. يمكن أن يفسر انخفاض نشاط النواة تحت المهاد، على ما يبدو، الحركات الرقصية التي تحدث في مرض هنتنغتون. من المعروف منذ فترة طويلة أن الآفات البؤرية للنواة تحت المهاد يمكن أن تسبب الرقص. من المرجح أن يكون فقدان عصبونات GABA والمادة P الممتدّة إلى الجزء الشبكي من المادة السوداء مسؤولاً عن اضطرابات حركة العين المُلاحظة في داء هنتنغتون. عادةً ما يُثبّط هذا المسار عصبونات الجزء الشبكي من المادة السوداء الممتدّة إلى التلة العلوية، والتي تُنظّم بدورها حركات العين السريعة. في داء هنتنغتون لدى الأطفال، تتأثر المسارات المذكورة أعلاه بشكل أشدّ، بالإضافة إلى فقدان مُبكّر لامتدادات الجسم المُخطّط إلى الجزء الداخلي من الكرة الشاحبة.
يوجد بروتين هنتنغتين، المُشفّر بواسطة الجين الذي تُسبّب طفرته مرض هنتنغتون، في تراكيب مختلفة من الدماغ وأنسجة أخرى. عادةً ما يوجد هنتنغتين بشكلٍ رئيسي في سيتوبلازم الخلايا العصبية. يوجد هذا البروتين في معظم الخلايا العصبية في الدماغ، إلا أن البيانات الحديثة تُظهر أن محتواه أعلى في الخلايا العصبية المصفوفية منه في الخلايا العصبية الستريوزومية، وأعلى في الخلايا العصبية الإسقاطية منه في الخلايا العصبية البينية. وبالتالي، ترتبط الحساسية الانتقائية للخلايا العصبية بمحتواها من هنتنغتين، والذي يوجد عادةً في بعض المجموعات العصبية.
كما هو الحال في أدمغة مرضى داء هنتنغتون، يُشكّل بروتين هنتنغتون في الفئران المُعدّلة وراثيًا للجزء الطرفي الأميني من جين داء هنتنغتون ذي عدد مُوسّع من التكرارات، تكتلات كثيفة في نوى الخلايا العصبية. تتشكل هذه التراكمات داخل النواة في الخلايا العصبية الإسقاطية المخططية (ولكن ليس في الخلايا العصبية البينية). في الفئران المُعدّلة وراثيًا، تتشكل هذه التراكمات قبل ظهور الأعراض بعدة أسابيع. تُشير هذه البيانات إلى أن بروتين هنتنغتون، الذي يحتوي على عدد مُتزايد من بقايا الجلوتامين التي تُشفّر تَشويهاتها تكرارات ثلاثية النوكليوتيدات، أو جزء منها، يتراكم في النواة، وقد يُضعف بالتالي تحكمها في الوظائف الخلوية.
أعراض مرض هنتنغتون
يصعب تحديد عمر ظهور الأعراض الأولى لدى مرضى داء هنتنغتون بدقة، نظرًا لظهور المرض تدريجيًا. قد تحدث تغيرات في الشخصية والسلوك، واضطرابات تنسيق خفيفة، قبل سنوات عديدة من ظهور الأعراض الأكثر وضوحًا. عند تشخيص المرض، يعاني معظم المرضى من حركات رقصية، وضعف في تنسيق الحركات الدقيقة، وبطء في حركات العين السريعة الإرادية. مع تقدم المرض، تضعف القدرة على تنظيم الأنشطة، وتضعف الذاكرة، ويصبح الكلام صعبًا، وتزداد اضطرابات حركة العين، ويضعف أداء الحركات المنسقة. على الرغم من عدم حدوث تغيرات في العضلات أو وضعية الجسم في المرحلة المبكرة من المرض، إلا أنه مع تقدم المرض، قد تتطور أوضاع خلل التوتر العضلي، والتي قد تتحول مع مرور الوقت إلى عرض سائد. في المرحلة المتأخرة، يصبح الكلام غير واضح، ويصعب البلع بشكل كبير، ويصبح المشي مستحيلًا. يتطور داء هنتنغتون عادةً على مدى 15-20 عامًا. في المرحلة النهائية، يكون المريض عاجزًا ويحتاج إلى رعاية مستمرة. النتيجة المميتة لا ترتبط بشكل مباشر بالمرض الأساسي، بل بمضاعفاته، على سبيل المثال، الالتهاب الرئوي.
الخرف في مرض هنتنغتون
رمز التصنيف الدولي للأمراض-10
P02.2. الخرف في مرض هنتنغتون (G10).
يتطور الخرف كأحد مظاهر عملية تنكسية ضامرة جهازية، مع تلف سائد في الجهاز المخططي للدماغ ونوى تحت المخ الأخرى. وهو وراثي بشكل جسمي سائد.
وكقاعدة عامة، يظهر المرض في العقد الثالث أو الرابع من العمر مع فرط الحركة الرقصية (وخاصة في الوجه والذراعين والكتفين والمشية)، وتغيرات الشخصية (أنواع من الشذوذ في الشخصية المثيرة والهستيرية والفصامية)، والاضطرابات الذهانية (الاكتئاب الخاص مع الكآبة والعبوس واضطراب المزاج؛ والمزاج البارانويدي).
من الأهمية بمكان للتشخيص الجمع بين فرط الحركة الرقصي والخرف والعبء الوراثي. وفيما يلي بيان خاص بهذا الخرف:
- التقدم البطيء (في المتوسط 10-15 سنة): الانفصال بين القدرة المتبقية على رعاية الذات وعدم الكفاءة الفكرية الواضحة في المواقف التي تتطلب عملاً عقليًا منتجًا (التفكير المفاهيمي، وتعلم أشياء جديدة)؛
- عدم انتظام واضح في الأداء العقلي، والذي يعتمد على اضطرابات واضحة في الانتباه وعدم ثبات مواقف المريض (التفكير "المتشنج"، المشابه لفرط الحركة)؛
- عدم انتظام الانتهاكات الواضحة للوظائف القشرية العليا؛
- العلاقة عكسية بين زيادة الخرف وشدة الاضطرابات الذهانية.
مع الأخذ بعين الاعتبار النسبة العالية من الاضطرابات الذهانية (أوهام الغيرة والاضطهاد) والاضطرابات المزعجة في الصورة السريرية للمرض، يتم العلاج باستخدام مضادات الذهان المختلفة التي تمنع مستقبلات الدوبامين (مشتقات الفينوثيازين والبيوتيروفينون) أو تقلل من مستوى الدوبامين في الأنسجة (ريزيربين).
يُستخدم هالوبيريدول (٢-٢٠ ملغ/يوم)، وتيابريد (١٠٠-٦٠٠ ملغ/يوم) لمدة لا تزيد عن ثلاثة أشهر، وثيوريدازين (حتى ١٠٠ ملغ/يوم)، وريزيربين (٠.٢٥-٢ ملغ/يوم)، ومضاد الاختلاج كلونازيبام (١.٥-٦ ملغ/يوم). تساعد هذه الأدوية على تقليل فرط الحركة، وتخفيف التوتر العاطفي، وتعويض اضطرابات الشخصية.
يُجرى علاج الاضطرابات النفسية في العيادات الداخلية مع مراعاة المتلازمة السائدة، وعمر المريض، وحالته العامة. أما في العيادات الخارجية، فتُطبّق مبادئ العلاج نفسها (العلاج المداوم المستمر لاضطرابات الحركة، والتغيير الدوري للدواء). وتُستخدم جرعات أقل من مضادات الذهان في العيادات الخارجية.
تشمل تدابير إعادة التأهيل للخرف الخفيف والمتوسط العلاج المهني، والعلاج النفسي، والتدريب المعرفي. من الضروري التعاون مع أفراد الأسرة وتقديم الدعم النفسي لمن يرعون المريض. تتمثل الطريقة الرئيسية للوقاية من المرض في الاستشارة الطبية والوراثية لأقرب أقارب المريض، مع إحالته إلى تحليل الحمض النووي عند اتخاذ قرار الإنجاب.
التشخيص غير مُرضٍ عمومًا. يتطور المرض ببطء، وعادةً ما يُؤدي إلى الوفاة بعد ١٠-١٥ عامًا.
[ 18 ]
ما الذي يزعجك؟
علاج مرض هنتنغتون
علاج داء هنتنغتون هو علاج عرضي. يمكن تثبيط الرقص والهياج جزئيًا باستخدام مضادات الذهان (مثل الكلوربرومازين بجرعة 25-300 ملغ فمويًا 3 مرات يوميًا، أو هالوبيريدول بجرعة 5-45 ملغ فمويًا مرتين يوميًا)، أو ريزيربين بجرعة 0.1 ملغ فمويًا مرة واحدة يوميًا. تُزاد الجرعات إلى أقصى حد يمكن تحمله (قبل ظهور الآثار الجانبية، مثل النعاس ومرض باركنسون؛ أما ريزيربين، فيُحتمل انخفاض ضغط الدم). يهدف العلاج التجريبي إلى تقليل انتقال الغلوتامات عبر مستقبلات Nmethyl-O-aspartate والحفاظ على إنتاج الطاقة في الميتوكوندريا. العلاج الذي يهدف إلى زيادة حمض غاما أمينوبوتيريك (GABA) في الدماغ غير فعال.
يُعدّ الفحص والاستشارات الجينية أمرًا بالغ الأهمية، إذ تظهر أعراض المرض بعد سنوات الإنجاب. يُحال الأشخاص الذين لديهم تاريخ عائلي إيجابي، وكذلك المهتمون بالفحص، إلى مراكز متخصصة، مع مراعاة جميع الآثار الأخلاقية والنفسية.
العلاج العرضي لمرض هنتنغتون
لا يوجد علاج فعال يوقف تطور داء هنتنغتون. أُجريت تجارب عديدة على أدوية مختلفة، ولكن لم يُحقق أي تأثير يُذكر. تُستخدم مضادات الذهان ومضادات مستقبلات الدوبامين الأخرى على نطاق واسع لتصحيح الاضطرابات النفسية والحركات اللاإرادية لدى مرضى داء هنتنغتون. تعكس الحركات اللاإرادية اختلالًا في التوازن بين نظامي الدوبامين وجابائيريك. بناءً على ذلك، تُستخدم مضادات الذهان لتقليل النشاط الدوباميني الزائد. ومع ذلك، يمكن أن تُسبب هذه الأدوية نفسها آثارًا جانبية إدراكية وخارج هرمية كبيرة. بالإضافة إلى ذلك، باستثناء الحالات التي يُصاب فيها المريض بالذهان أو الهياج، لم تُثبت فعاليتها. غالبًا ما تُسبب مضادات الذهان عسر البلع أو اضطرابات الحركة الأخرى أو تُفاقمها. قد تكون مضادات الذهان من الجيل الأحدث، مثل الريسبيريدون والكلوزابين والأولانزابين، مفيدة بشكل خاص في علاج داء هنتنغتون لأنها تُسبب آثارًا جانبية خارج هرمية أقل، ولكنها قد تُخفف أعراض جنون العظمة أو زيادة التهيج.
يُقلل تيترابينازين وريزيربين أيضًا من نشاط الجهاز الدوباميني، ويمكنهما تخفيف شدة الحركات اللاإرادية في المراحل المبكرة من المرض. ومع ذلك، قد تُسبب هذه الأدوية الاكتئاب. ولأن المرض نفسه غالبًا ما يُسبب الاكتئاب، فإن هذا الأثر الجانبي يحدّ بشكل كبير من استخدام ريزيربين وتيترابينازين. في المراحل المتأخرة من المرض، تموت الخلايا الحاملة لمستقبلات الدوبامين، مما يُضعف أو يُفقد فعالية مضادات مستقبلات الدوبامين.
تُستخدم مضادات الذهان ومضادات الاكتئاب ومضادات القلق لعلاج الذهان والاكتئاب والانفعال لدى مرضى داء هنتنغتون، ولكن يجب وصفها فقط طالما استمرت هذه الأعراض. الأدوية التي قد تكون مفيدة في مرحلة ما من المرض قد تصبح غير فعالة أو حتى ضارة مع تقدم المرض.
تم اختبار مُنشِّطات مستقبلات GABA لدى مرضى داء هنتنغتون، حيث ثبت أن داء هنتنغتون يُسبب انخفاضًا ملحوظًا في مستويات GABA في الجسم المخطط، بالإضافة إلى فرط حساسية مستقبلات GABA في مناطق إسقاطه. أثبتت البنزوديازيبينات فعاليتها في الحالات التي تتفاقم فيها الحركات اللاإرادية والضعف الإدراكي بسبب التوتر والقلق. يجب وصف جرعات منخفضة من هذه الأدوية لتجنب الآثار المهدئة غير المرغوب فيها. في معظم مرضى داء هنتنغتون، لا يُؤدي أيٌّ من هذه الأدوية إلى تحسن كبير في جودة الحياة.
في حالات داء هنتنغتون المبكر المصحوب بأعراض باركنسون، قد تُجرَّب عوامل الدوبامين، لكن فعاليتها محدودة. علاوة على ذلك، قد يُسبِّب ليفودوبا الرمع العضلي أو يزيده لدى هؤلاء المرضى. في الوقت نفسه، قد يُخفِّف الباكلوفين التصلب لدى بعض مرضى داء هنتنغتون.
[ 26 ]، [ 27 ]، [ 28 ]، [ 29 ]
العلاج الوقائي (العصبي) لمرض هنتنغتون
على الرغم من أن الخلل الجيني في مرض هنتنغتون معروف، إلا أن كيفية إسهامه في التنكس العصبي الانتقائي لا تزال غير واضحة. يُفترض أن العلاجات الوقائية التي تهدف إلى تقليل الإجهاد التأكسدي والسمية الإثارية قد تُبطئ أو تُوقف تطور المرض. قد يكون الوضع مشابهًا إلى حد ما للتنكس الكبدي العدسي، حيث ظل الخلل الجيني مجهولًا لسنوات عديدة، ولكن العلاجات الوقائية التي تهدف إلى معالجة التأثير الثانوي، تراكم النحاس، قد شُفيت. في هذا الصدد، حظيت فرضية ارتباط مرض هنتنغتون باضطراب في استقلاب الطاقة وموت الخلايا بسبب السمية الإثارية باهتمام خاص. قد يُسبب المرض نفسه موت الخلايا بسبب التراكم داخل النواة لشظايا هنتنغتين الطرفية الأمينية، مما يُعطل الوظائف الخلوية والأيضية. قد تؤثر هذه العملية على بعض مجموعات الخلايا العصبية بدرجة أكبر من غيرها نظرًا لحساسيتها العالية للتلف الناتج عن السمية الإثارية. في هذه الحالة، يُمكن للعلاج الوقائي بمضادات مستقبلات الأحماض الأمينية المُثيرة أو العوامل التي تمنع تلف الجذور الحرة أن يمنع أو يُؤخر ظهور المرض وتطوره. في النماذج المخبرية لمرض التصلب الجانبي الضموري، تبيّن أن مضادات الأكسدة ومضادات المستقبلات (RAAs) قادرة على إبطاء تطور المرض. وقد تكون أساليب مماثلة فعّالة في علاج داء هنتنغتون. وتُجرى حاليًا تجارب سريرية على مضادات مستقبلات الغلوتامات وعوامل تُعزز وظيفة المركب الثاني من سلسلة نقل الإلكترونات في الميتوكوندريا.
[ 30 ]، [ 31 ]، [ 32 ]، [ 33 ]، [ 34 ]، [ 35 ]، [ 36 ]، [ 37 ]، [ 38 ]، [ 39 ]