خبير طبي في المقال

منشورات جديدة
مرض شاركو-ماري-ماري-توث.
آخر مراجعة: 12.07.2025

تتم مراجعة جميع محتويات iLive طبياً أو التحقق من حقيقة الأمر لضمان أكبر قدر ممكن من الدقة الواقعية.
لدينا إرشادات صارمة من مصادرنا ونربط فقط بمواقع الوسائط ذات السمعة الطيبة ، ومؤسسات البحوث الأكاديمية ، وطبياً ، كلما أمكن ذلك استعراض الأقران الدراسات. لاحظ أن الأرقام الموجودة بين قوسين ([1] و [2] وما إلى ذلك) هي روابط قابلة للنقر على هذه الدراسات.
إذا كنت تشعر أن أيًا من المحتوى لدينا غير دقيق أو قديم. خلاف ذلك مشكوك فيه ، يرجى تحديده واضغط على Ctrl + Enter.
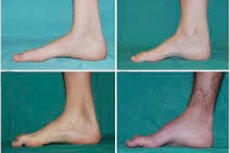
ضمور العضلات الشظوية، أو متلازمة أو مرض شاركو ماري توث، هو مجموعة من الأمراض الوراثية المزمنة التي تسبب تلف الأعصاب الطرفية.
وفقًا للتصنيف الدولي للأمراض (ICD-10)، في قسم أمراض الجهاز العصبي، يُرمز لهذا المرض بالرمز G60.0 (اعتلال عصبي حركي وحسي وراثي). وهو مُدرج أيضًا في قائمة الأمراض النادرة.
علم الأوبئة
وفقًا للإحصائيات السريرية، يبلغ معدل انتشار جميع أنواع مرض شاركو ماري توث لكل 100 ألف من السكان 19 حالة (وفقًا لمصادر أخرى، حالة واحدة لكل 2.5-10 آلاف من السكان).
يُمثل النوع الأول من CMT حوالي ثلثي الحالات (حالة واحدة لكل 5000 إلى 7000 نسمة)، ويرتبط ما يقرب من 70% منها بتكرار جين PMP22. يُعاني أكثر من 1.2 مليون شخص حول العالم من هذا النوع من المرض.
يُقدر معدل الإصابة بمرض شاركو-ماري-توث من النوع الرابع بحوالي 1-5 حالات لكل 10000 طفل. [ 1 ]
الأسباب مرض شاركو-ماري-ماري-توث
وفقًا لتصنيف المتلازمات العصبية المتعددة ، فإن ضمور العضلات الشظوية (الشظوية)، أو ضمور الأعصاب شاركو-ماري-توث أو مرض شاركو-ماري-توث (المختصر باسم CMT) يشير إلى اعتلالات عصبية حركية حسية محددة وراثيًا. [ 2 ]
أي أن أسباب حدوثه هي الطفرات الجينية. وحسب طبيعة الانحرافات الجينية، تُميّز الأنواع الرئيسية لهذه المتلازمة: متلازمة إزالة الميالين ومتلازمة المحور العصبي. تشمل المجموعة الأولى متلازمة شاركو-ماري-توث من النوع الأول (CMT1)، التي تحدث بسبب تضاعف جين PMP22 على الكروموسوم 17، الذي يُشفّر بروتين الميالين المحيطي عبر الغشاء 22. ونتيجةً لذلك، يحدث إزالة الميالين القطعية لغلاف المحور العصبي (عمليات الخلية العصبية) وانخفاض في سرعة توصيل الإشارات العصبية. بالإضافة إلى ذلك، قد تحدث طفرات في بعض الجينات الأخرى.
الشكل المحوري هو مرض شاركو-ماري-توث من النوع الثاني (CMT2)، الذي يؤثر على المحاور العصبية نفسها ويرتبط بتغيرات مرضية في جين MFN2 في الموضع 1p36.22، الذي يُشفّر بروتين الغشاء ميتوفوسين-2، الضروري لاندماج الميتوكوندريا وتكوين شبكات الميتوكوندريا الوظيفية داخل الخلايا العصبية الطرفية. يوجد أكثر من اثني عشر نوعًا فرعيًا من CMT2 (مع طفرات في جينات محددة).
تجدر الإشارة إلى أنه تم تحديد أكثر من مئة جين حاليًا، يتسبب تلفها، الذي ينتقل بالوراثة، في أنواع فرعية مختلفة من مرض شاركو-ماري-توث. على سبيل المثال، تؤدي الطفرات في جين RAB7 إلى الإصابة بمرض شاركو-ماري-توث من النوع 2B؛ ويؤدي تغيير جين SH3TC2 (الذي يُشفّر أحد بروتينات غشاء خلايا شوان) إلى الإصابة بمرض شاركو-ماري-توث من النوع 4C، والذي يظهر في مرحلة الطفولة، ويتميز بزوال الميالين في الخلايا العصبية الحركية والحسية (هناك اثني عشر شكلًا ونصف من النوع 4 من هذا المرض).
يبدأ النوع الثالث النادر من مرض شاركومورف-ماري (CMT) (يسمى متلازمة ديجيرين-سوتاس) في التطور في مرحلة الطفولة المبكرة ويحدث بسبب طفرات في جينات PMP22 وMPZ وEGR2 وغيرها من الجينات.
عندما يحدث CMT من النوع 5 في سن 5-12 سنة، لا يتم ملاحظة الاعتلال العصبي الحركي فقط (في شكل شلل تشنجي في الأطراف السفلية)، ولكن أيضًا تلف الأعصاب البصرية والسمعية.
يعد ضعف العضلات وضمور العصب البصري (مع فقدان البصر) بالإضافة إلى مشاكل التوازن من الأعراض النموذجية لمرض شاركو-ماري-توث من النوع السادس. وفي مرض شاركو-ماري-توث من النوع السابع لا يوجد اعتلال عصبي حركي وحسي فحسب، بل يوجد أيضًا مرض في الشبكية في شكل التهاب الشبكية الصباغي.
أكثر شيوعًا بين الذكور، مرض CMT المرتبط بالكروموسوم X أو مرض شاركو ماري توث مع شلل رباعي الأطراف (ضعف حركة كل من الذراعين والساقين) هو نوع مزيل للميالين ويُعتقد أنه ينتج عن طفرة في جين GJB1 على الذراع الطويلة للكروموسوم X، والذي يشفر الكونيكسين 32، وهو بروتين عبر غشائي لخلايا شوان والخلايا القليلة التغصنات ينظم انتقال الإشارات العصبية. [ 3 ]
عوامل الخطر
عامل الخطر الرئيسي للإصابة بمرض CMT هو التاريخ العائلي للمرض، أي في الأقارب المقربين.
وفقًا لعلماء الوراثة، إذا كان كلا الوالدين حاملين للجين المتنحي الجسدي لمرض شاركو-ماري-توث، فإن خطر إنجاب طفل مصاب بهذا المرض يبلغ 25%. ويُقدر خطر أن يكون الطفل حاملًا لهذا الجين (دون ظهور أي أعراض) بنسبة 50%.
في حالة الوراثة المرتبطة بالكروموسوم X (عندما يكون الجين المتحور على الكروموسوم X للمرأة)، هناك خطر بنسبة 50% أن تنقل الأم الجين إلى ابنها، الذي سيُصاب بمرض شاركو-ماري توث (CMT). قد لا يُصاب الطفل الأنثى بالمرض، ولكن قد يرث أبناء الابنة (الأحفاد) الجين المعيب، ويتطور المرض.
طريقة تطور المرض
في أي نوع من أنواع مرض شاركو-ماري-توث، يحدث المرض بسبب خلل وراثي في الأعصاب الطرفية: الحركية (الحركية) والحسية (الحساسة).
إذا كان نوع CMT هو مزيل للميالين، فإن تدمير أو خلل غمد الميالين الذي يحمي محاور الأعصاب الطرفية يؤدي إلى تباطؤ في انتقال النبضات العصبية في الجهاز العصبي الطرفي - بين الدماغ والعضلات والأعضاء الحسية.
في النوع المحوري من المرض تتأثر المحاور نفسها، مما يؤثر سلبًا على قوة الإشارات العصبية، والتي لا تكفي لتحفيز العضلات والأعضاء الحسية بشكل كامل.
اقرأ أيضًا:
كيف تنتقل متلازمة شاركو-ماري-توث؟ يمكن أن تُورث الجينات المعيبة بطريقة جسمية سائدة أو جسمية متنحية.
النوع الأكثر شيوعًا، وهو الوراثة الجسدية السائدة، يحدث عندما تكون هناك نسخة واحدة من الجين المتحور (يحملها أحد الوالدين). ويُقدر احتمال انتقال مرض شاركو-ماري-توث إلى كل طفل مولود بنسبة 50%. [ 4 ]
في حالة الوراثة الجسدية المتنحية، هناك حاجة إلى نسختين من الجين المعيب (نسخة واحدة من كل والد لا تظهر عليه أي علامات للمرض) حتى يتطور المرض.
في 40-50% من الحالات، يحدث إزالة الميالين الوراثية السائدة جسميًا، أي CMT من النوع 1؛ وفي 12-26% من الحالات، يحدث CMT محوري، أي النوع 2. وفي 10-15% من الحالات، يُلاحظ الوراثة المرتبطة بالكروموسوم X. [ 5 ]
الأعراض مرض شاركو-ماري-ماري-توث
عادةً ما تبدأ الأعراض الأولى لهذا المرض بالظهور في مرحلتي الطفولة والمراهقة، وتتطور تدريجيًا طوال الحياة، مع أن المتلازمة قد تظهر لاحقًا. تتفاوت مجموعة الأعراض، ويستحيل التنبؤ بمعدل تطور المرض أو شدته.
تشمل الأعراض النموذجية للمرحلة الأولية زيادة التعب العام؛ وضعف عضلات القدمين والكاحلين والساقين؛ وضعف ردود الفعل. هذا يُصعّب حركة القدم ويؤدي إلى خلل في المشية (اضطراب في المشية) على شكل ارتفاع شديد في الساقين، وغالبًا ما يكون مصحوبًا بالتعثر والسقوط المتكرر. قد تشمل علامات مرض شاركو-ماري-توث لدى الأطفال الصغار خرقًا واضحًا وصعوبات في المشي غير معتادة على العمر، مصحوبة بسقوط القدمين. كما تُعد تشوهات القدم من السمات المميزة: ارتفاع قوس القدم (القدم المجوفة) أو تسطح القدم الشديد، وانحناء أصابع القدم (المطرقة).
في حالة المشي على أصابع القدم على خلفية انخفاض التوتر العضلي، قد يشتبه طبيب الأعصاب في أن الطفل مصاب بمرض شاركو ماري تشامبرز من النوع الرابع، حيث قد يكون الأطفال غير قادرين على المشي بحلول سن المراهقة.
مع تطور المرض، ينتشر ضمور العضلات وضعفها إلى الأطراف العلوية، مما يُصعّب أداء المهارات الحركية الدقيقة والمهام اليدوية العادية. ويُشير انخفاض الإحساس باللمس والقدرة على الشعور بالحرارة والبرودة، بالإضافة إلى خدر في القدمين واليدين، إلى تلف في محاور الأعصاب الحسية.
في مرض شاركو ماري توث من النوعين 3 و 6 الذي يظهر في مرحلة الطفولة، يتم ملاحظة الترنح الحسي (ضعف تنسيق الحركات والتوازن)، وارتعاش العضلات والرعشة، وتلف العصب الوجهي، وضمور العصب البصري مع الرجفة، وفقدان السمع.
وفي المراحل اللاحقة، قد يكون هناك اهتزاز لا يمكن السيطرة عليه (رعشة) وتشنجات عضلية متكررة؛ ويمكن أن تؤدي مشاكل الحركة إلى تطور الألم: العضلي، والمفاصل، والأعصاب.
المضاعفات والنتائج
يمكن أن يكون لمرض شاركو-ماري-توث مضاعفات وعواقب مثل:
- زيادة تكرار الالتواءات والكسور؛
- التقلصات المرتبطة بتقصير العضلات والأوتار المحيطة بالمفصل؛
- الجنف (انحناء العمود الفقري)؛
- مشاكل في التنفس - عندما تتضرر الألياف العصبية التي تغذي عضلات الحجاب الحاجز:
- فقدان القدرة على الحركة بشكل مستقل.
التشخيص مرض شاركو-ماري-ماري-توث
يتضمن التشخيص الفحص السريري، والتاريخ المرضي (بما في ذلك التاريخ العائلي)، والفحص العصبي والجهازي.
تُجرى اختبارات للتحقق من مدى الحركة والحساسية وردود فعل الأوتار. ويمكن تقييم التوصيل العصبي باستخدام التشخيصات الآلية - تخطيط كهربية العضل أو تخطيط كهربية العصب العضلي. وقد يلزم أيضًا إجراء تصوير بالموجات فوق الصوتية أو بالرنين المغناطيسي. [ 6 ]
إن إجراء فحوصات جينية أو فحوصات الحمض النووي للكشف عن الطفرات الجينية الأكثر شيوعًا المسببة لمرض شاركو-ماري-توث في عينة دم أمر محدود، نظرًا لعدم توفر فحوصات الحمض النووي حاليًا لجميع أنواع المرض. لمزيد من المعلومات، راجع قسم الفحوصات الجينية.
في بعض الحالات يتم إجراء خزعة من العصب الطرفي (عادة العصب الظنبوبي).
تشخيص متباين
يتضمن التشخيص التفريقي اعتلالات عصبية محيطية أخرى، وضمور دوشين العضلي، ومتلازمات النخاع والوهن العضلي، والاعتلال العصبي السكري، واعتلالات النخاع في التصلب الجانبي المتعدد والضموري، ومتلازمة غيلان باريه، والصدمات التي تصيب العصب الشظوي وضموره (بما في ذلك عند الضغط عليه بين الأقراص القطنية للعمود الفقري)، وتلف المخيخ أو المهاد، بالإضافة إلى الآثار الجانبية للعلاج الكيميائي (أثناء العلاج بالعقاقير المضادة للخلايا مثل فينكريستين أو باكليتاكسيل). [ 7 ]
من الاتصال؟
علاج او معاملة مرض شاركو-ماري-ماري-توث
يشمل علاج هذا المرض الوراثي اليوم العلاج بالتمارين الرياضية (التي تهدف إلى تقوية وتمديد العضلات)؛ والعلاج المهني (الذي يساعد المرضى الذين يعانون من ضعف عضلات اليدين)؛ واستخدام الأجهزة التقويمية لتسهيل المشي. وعند الضرورة، تُوصف مسكنات الألم أو مضادات الاختلاج. [ 8 ]
في حالات القدم المسطحة الشديدة، يمكن إجراء قطع العظم، وفي حالة تشوه الكعب، يوصى بالتصحيح الجراحي - تثبيت المفصل. [ 9 ]
تُجرى أبحاثٌ حول العامل الوراثي للمرض وطرق علاجه. ولم يُسفر استخدام الخلايا الجذعية، أو بعض الهرمونات، أو الليسيثين، أو حمض الأسكوربيك عن نتائج إيجابية حتى الآن.
لكن بفضل الأبحاث الحديثة، قد يظهر في المستقبل القريب علاج جديد لمرض شاركو-ماري-توث. فمنذ عام ٢٠١٤، تعمل شركة فارنيكست الفرنسية على تطوير دواء PXT3003، ومنذ منتصف عام ٢٠١٩، تُجرى تجارب سريرية على هذا الدواء لعلاج النوع الأول من CMT لدى البالغين، والذي يُثبط التعبير المتزايد لجين PMP22، ويُحسّن عملية تغميد الأعصاب الطرفية، ويُخفف الأعراض العصبية العضلية.
يعمل متخصصون من شركة ساريبتا ثيرابيوتكس الطبية (الولايات المتحدة الأمريكية) على تطوير علاج جيني لمرض شاركو-ماري-توث من النوع الأول. سيستخدم هذا العلاج فيروسًا غديًا غير ضار (AAV) من جنس ديبيندوفيروس، ذو جينوم خطي أحادي السلسلة من الحمض النووي، والذي سينقل جين NTF3 إلى الجسم، مشفّرًا بروتين نيوروتروفين-3 (NT-3) الضروري لعمل خلايا شوان العصبية.
ستبدأ شركة Helixmith التجارب السريرية للعلاج الجيني الذي طورته كوريا الجنوبية Engensis (VM202) بحلول نهاية عام 2020 لعلاج أعراض العضلات في النوع الأول من مرض شاركو-ماري توث. [ 10 ]
الوقاية
يمكن الوقاية من مرض شاركو-ماري توث (CMT) من خلال الاستشارة الوراثية للوالدين المستقبليين، خاصةً إذا كان لأحد الزوجين تاريخ عائلي للإصابة بالمرض. ومع ذلك، فقد تم تحديد حالات طفرات جينية نقطية جديدة، أي في غياب المرض في التاريخ العائلي.
خلال فترة الحمل، يمكن التحقق من احتمالية إصابة الطفل المستقبلي بمرض شاركو ماري توث عن طريق خزعة الزغابات المشيمية (من 10 إلى 13 أسبوعًا من الحمل)، بالإضافة إلى تحليل السائل الأمنيوسي (في 15-18 أسبوعًا).
توقعات
بشكل عام، يعتمد تشخيص أنواع مختلفة من مرض شاركو-ماري-توث على شدته السريرية، ولكن في جميع الحالات، يتطور المرض ببطء. يعاني العديد من المرضى من إعاقات، وإن كان هذا لا يقلل من متوسط العمر المتوقع.