
متلازمة انجلمان عند الأطفال والبالغين
آخر مراجعة: 23.04.2024

تتم مراجعة جميع محتويات iLive طبياً أو التحقق من حقيقة الأمر لضمان أكبر قدر ممكن من الدقة الواقعية.
لدينا إرشادات صارمة من مصادرنا ونربط فقط بمواقع الوسائط ذات السمعة الطيبة ، ومؤسسات البحوث الأكاديمية ، وطبياً ، كلما أمكن ذلك استعراض الأقران الدراسات. لاحظ أن الأرقام الموجودة بين قوسين ([1] و [2] وما إلى ذلك) هي روابط قابلة للنقر على هذه الدراسات.
إذا كنت تشعر أن أيًا من المحتوى لدينا غير دقيق أو قديم. خلاف ذلك مشكوك فيه ، يرجى تحديده واضغط على Ctrl + Enter.

هناك عدد من الأمراض التي تكون فيها التعبيرات مثل "اعتني بنفسك ولا تمرض" سليمة ، على الأقل ، سخيفة. هذه الأمراض ، التي تكون فيها بعض التشوهات العقلية والجسدية جزءا لا يتجزأ من جسم الطفل حتى قبل الولادة ، ولكن الآباء ليس لديهم أي شعور بالذنب. تحدث هذه الأمراض بسبب طفرات أو اضطرابات في مجموعات الكروموسومات وتسمى بالكروموسومات أو الجينات. متلازمة انجلمان ، متلازمة داون ، Patau ، Edwards ، Turner ، Prader-Willi ليست سوى جزء من الأمراض الوراثية من قائمة لائقة إلى حد ما.
متلازمة شخص سعيد
هذه المرة نحن نتحدث عن هذا المرض، الذي سمي على اسم طبيب الأطفال البريطاني هاري Angelman من أن أول أثار قضية عام 1965 مشكلة، واجه عشية ممارسته مع ثلاثة أطفال غير عادية، وتوحدها أعراض غريبة المشتركة. سمى الطبيب هؤلاء الأطفال الأطفال الدمى وكتب عنهم مقالا كان يسمى في الأصل "الأطفال العرائس". وكتب المقال نفسه واسمه تحت انطباع صورة في أحد المتاحف في فيرونا. صورت الصورة صبيًا ضاحكًا ، وكان يطلق عليه "بوي-دمية". جمعية هو مبين في الصورة الطفل مع الأطفال الثلاثة مع Angelman التي واجهت مرة واحدة في ممارساتها، ودفع الأطفال طبيب أطفال تجمع في مجموعة واحدة بسبب حالة القائمة.
حقيقة أن الأطفال لاحظت في المادة لم تكن لاحظت من قبل الأطباء الآخرين ليس من المستغرب. بعد كل شيء ، للوهلة الأولى يبدو أن لديهم أمراض مختلفة تماما ، لذلك اختلفت الصورة السريرية العامة للمرض في 3 حالات مختلفة. قد يكون علم الأمراض الكروموسومات "الجديدة" محل اهتمام علماء آخرين ، ولكن في ذلك الوقت لم يكن علم الوراثة قد تطور بعد لتأكيد فرضية الطبيب الإنجليزي. لذلك ، تم التخلي عن المادة بعد اهتمام معين في ذلك لفترة طويلة إلى فوج بعيد.
إن الإشارة التالية إلى متلازمة أنجلمان ، وهكذا سميت الآن بمقال طبيب الأطفال من إنجلترا ، جي أنجليمان ، تعود إلى بداية الثمانينيات من القرن العشرين. وفقط في عام 1987 كان من الممكن العثور على السبب وراء ولادة جزء صغير من الأطفال مع مثل هذه الانحرافات ، والتي من الجانب تبدو مبتسمة وسعيدة باستمرار. في الواقع ، هذا ليس كذلك ، والابتسامة مجرد كآبة ، وراءها النفس البشرية غير السعيدة وآلام الوالدين.
علم الأوبئة
الطفرة الصبغية في الطفل ، وفقا للإحصاءات ، يمكن أن تتطور على خلفية هذه الطفرات في الوالدين ، وفي غياب مثل هذا. لا توجد شخصية وراثية واضحة في متلازمة انجلمان (SA) ، ولكن احتمال تطور علم الأمراض لدى الآباء الذين لديهم طفرات صبغية مرتفع للغاية.
ومن المثير للاهتمام أيضًا أنه إذا كان لدى عائلة بالفعل طفل مصاب بالتهاب الأمعاء ، فهناك فرصة بنسبة واحد في المائة لوجود طفل ثان من النوع نفسه ، حتى إذا كان الوالدان يتمتعان بصحة جيدة.
لا توجد حتى الآن إحصاءات دقيقة عن عدد المرضى الذين يعانون من متلازمة أنجلمان. ربما يكون الخطأ هو مجموعة متنوعة من الأعراض التي يمكن أن تحدث في تكوين معين أو لفترة طويلة لا تنشأ على الإطلاق. من المفترض أن معدل انتشار المرض هو: طفل واحد لكل 20.000 وليد. لكن هذا الرقم تقريبي جدا.
الأسباب متلازمة انجلمان
متلازمة انجلمان هي الاسم الطبي لعلم الأمراض الصبغية ، لكنها ليست الوحيدة بأي حال من الأحوال. في الناس يسمى هذا المرض أيضا متلازمة الأطفال العرائس ، ومتلازمة دمية سعيدة ، ومتلازمة Petrushka ، ومتلازمة دمية تضحك. نعم أي نوع من الأسماء لا يستطيع الناس أن يأتوا بها (في بعض الأحيان إهانة للمرضى أنفسهم وأولياء أمورهم) ، لكن المرض مرض ، بغض النظر عن مدى ضحكته الظاهرة ومهما كانت الأسباب.
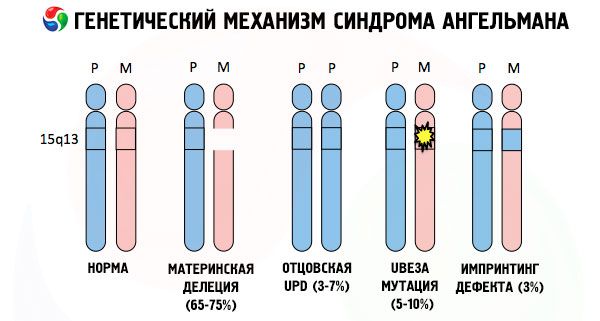
وأسباب تطور متلازمة أنجلمان ، بالإضافة إلى العديد من الأمراض الوراثية الأخرى ، هي في جميع الحالات انتهاكات في بنية أحد الكروموسومات أو مجموعة الكروموسوم ككل. ولكن في حالتنا فقط ، تكمن المشكلة برمتها في الكروموسومات الخمسة عشر التي تنتقل من الأم. أي لا يوجد لدى الكروموسومات الأبوية في هذه الحالة أي انحرافات ، لكن الأنثى تخضع لبعض التحولات.
وفقا لنوع الشذوذ الكروموسومي ، تشير متلازمة انجلمان إلى الطفرات الصبغية. مثل هذه الطفرات هي:
- الحذف (غياب منطقة صبغي يحتوي على مجموعة محددة من الجينات ، إذا لم يكن أحد الجينات لا يشير إلى microdeletion) هو نتيجة لاثنين من الانقطاعات وموحدة لم الشمل ، عندما تضيع منطقة الكروموسوم الأصلي.
- الازدواجية (وجود موقع إضافي في الكروموسوم ، وهو نسخة من المتوفر بالفعل) ، والذي يؤدي في معظم الحالات إلى وفاة شخص ، أقل في كثير من الأحيان - إلى العقم.
- انقلاب (انقلاب من أقسام كروموسوم عند 180 درجة، أي في الاتجاه المعاكس، ومن ثم يتم ترتيب الجينات فيها بترتيب عكسي) عندما ينتهي كروموسوم كسر اتصال في ترتيب مختلف عن الأصلي.
- الإدراج (إذا كان جزء من المادة الوراثية في الكروموسوم ليس في مكانه) ،
- نقل (إذا كان جزء من الكروموسوم ينضم إلى كروموسوم آخر ، يمكن أن تكون هذه الطفرة متبادلة دون فقد المواقع).
الحصول على كروموسوم متحور من أم غير متأمّنة ، فالطفل محكوم عليه أن يولد مع الانحرافات مقدما. السبب الأكثر شيوعًا لتطور متلازمة أنغلمان لا يزال هو حذف الكروموسوم 15 من الأمهات ، عندما لا توجد منطقة صغيرة فيه. والطفرات الأقل شيوعًا في متلازمة "دمية الضحك" هي:
- النبات،
- ديوسوميا الأب الوحيد (إذا كان الطفل قد تلقى زوجًا من الكروموسومات من الأب ، فإن الكروموسوم الأم غير موجود)
- تحور الجينات في الحمض النووي ، وهي مادة البناء الرئيسية (الجينية) وتعليمات الاستخدام الصحيح لها (على وجه الخصوص ، تحور جين ube3a في الكروموسوم الأمومي).
وجود واحدة من هذه الطفرات في الآباء هو عامل خطر لمتلازمة أنجلمان في الأطفال. ولكن ليس فقط الطفرات الكروموسومية ، ولكن أيضا الطفرات الجينية (التي ترتبط بالتغير الكمي في مجموعات الكروموسومات وتوجد في كثير من الأحيان أكثر من الكروموسومات) يمكن أن تؤدي إلى تطور المرض في الطفل. يمكن أن يعزى إلى طفرات جينية مشتركة ثلاثة أنواع من الكروموسومات (إذا كان لدى الشخص مجموعة كروموسوم بها أكثر من 46 كروموسوم).
لا ينبغي بالضرورة أن يكون لوالدة الطفل آباء لديهم تشوهات صبغية. ومع ذلك هناك نسبة معينة من المرضى الذين يكون مرضهم وراثي.

طريقة تطور المرض
دعونا حفر قليلا في علم الأحياء ، على نحو أدق في علم الوراثة. وترد المعلومات الوراثية لكل فرد من جسم الإنسان في 23 زوجا من الكروموسومات. وينتقل كروموسوم واحد من هذا الزوج إلى الطفل من الأب ، والآخر من الأم. تختلف جميع أزواج الكروموسومات في الشكل والحجم وتحمل في حد ذاتها معلومات معينة. لذا ، فإن 23 زوجا من الكروموسومات (X و Y الصبغيات) مسؤولة عن تكوين الصفات الجنسية للطفل (XX - فتاة ، XY-boy ، في حين يمكن للطفل الحصول على كروموسوم Y من والده فقط).
من الناحية المثالية ، يتلقى الطفل من والديه 46 صبغيات ، والتي تشكل صفاته الوراثية ، وتحديده مسبقا كفرد. يسمى عدد أكبر من الكروموسومات بالتثلث الصبغي ويعتبر انحرافًا عن القاعدة. على سبيل المثال ، يؤدي وجود 47 كروموسوم في مجموعة الكروموسومات (النمط النووي ، الذي يحدد الأنواع والسمات الفردية) إلى ظهور متلازمة داون.
إذا كانت الصبغيات ملوّنة بصبغة خاصة ، فعندئذ في المجهر يمكن للمرء أن يرى عصابات من ظلال مختلفة على طول كل منها. داخل كل فرقة عدد ضخم من الجينات. يتم ترقيم جميع هذه النطاقات من قبل العلماء ولها موقع ثابت. يعتبر غياب أحد النطاقات انحرافًا عن القاعدة. مع متلازمة أنجلمان ، يمكنك في كثير من الأحيان ملاحظة غياب أجزاء من الكروموسوم الأمومي في الفترة الفاصلة q11-q13 الموجودة في الذراع الطويلة ، وهو عدد قواعد الدنا التي يبلغ عددها حوالي 4 ملايين فقط.
المكون الرئيسي للكروموسوم هو جزيء دنا طويل بشكل لا يصدق يحتوي على آلاف الجينات وعشرات ومئات الملايين من القواعد النيتروجينية. وهكذا ، فإن الكروموسومات الخمسة عشر المسؤولة عن تطوير متلازمة أنجلمان وعدة حالات أخرى ، تحتوي على 1200 جين وحوالي 100 مليون قاعدة. أي انتهاكات في بنية جزيء الحمض النووي سوف تؤثر بالضرورة على مظهر وتطور الطفل الذي لم يولد بعد.
يتم تحويل المعلومات الجينية الواردة في الجينات إلى البروتين أو الحمض النووي الريبي. هذه العملية تسمى التعبير الجيني. وبالتالي ، فإن المعلومات الوراثية الواردة من الآباء تتلقى شكلا ومضمونا ، يتجسد في وريثهم الفريد للجنس الأنثوي أو الذكوري.
وهناك عدد من الأمراض مع نوع غير الكلاسيكي من الميراث، بما في ذلك متلازمة أنجلمان، حيث الجينات التي وردت من أولياء الأمور كجزء من كروموزوم والآباء بصمة فريدة من نوعها وتعبر عن نفسها بطرق مختلفة.
لذا، متلازمة أنجلمان هي مثال ساطع على يطبع الجينومية، حيث التعبير عن الجينات في جسم الطفل اعتمادا مباشرا على ومنهم من الأم المستمدة الأليلات (أشكال مختلفة من نفس الجينات التي تم الحصول عليها من الأب والأم وتقع على أجزاء متطابقة من كروموزوم) . أي يؤدي إلى ظهور تشوهات متلازمة كروموسوم في الأمهات، في حين أن الطفرات واضطرابات الأب سبب هيكل كروموسوم أمراض مختلفة جدا.
في هذا المرض يكون هناك نقص الجينات المحددة في الكروموسوم الأمهات أو فقدان / انخفاض في نشاط الجينات الفردية (في معظم الحالات الجينات ube3a تشارك في عملية التمثيل الغذائي اليوبيكويتين - تدهور البروتين البروتينات التنظيمية الأخرى). ونتيجة لذلك ، يتم تشخيص الطفل مع تشوهات في النمو العقلي وتشوهات جسدية.
الأعراض متلازمة انجلمان
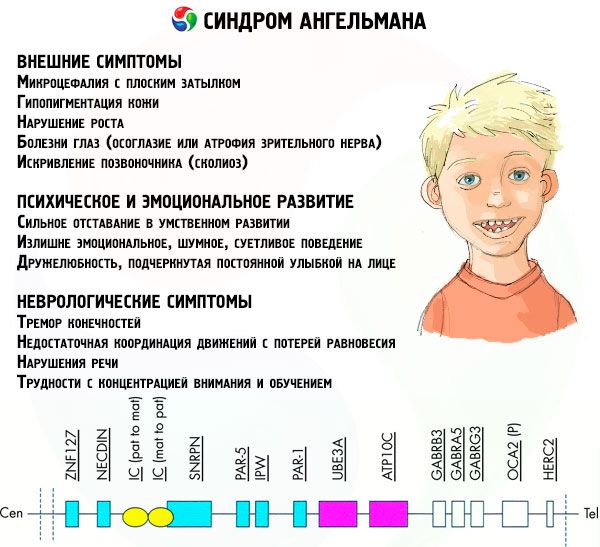
تؤثر أعراض متلازمة انجلمان على الجوانب المختلفة لحياة الطفل ونموه: الجسمي والعصبي والنفسي. بناء على ذلك ، يمكننا التمييز بين 3 مجموعات من الأعراض التي تشير إلى تطور هذه الحالة المرضية.
- الأعراض الخارجية أو الجسدية:
- رأس صغير بشكل غير متناسب مقارنة مع الجذع والأطراف ذات الحجم العادي ،
- فم واسع جدا ،
- على الوجه هناك دائما ابتسامة (مع فم مفتوح) ،
- أسنان نادرة
- الشفة العليا الضيقة ،
- في كثير من الأحيان يصرخ لسان عريض ،
- جاحظ الفك السفلي ،
- الذقن الحاد
- البشرة خفيفة جدا ، وغالبا ما يكون الشعر (المهق ، يرتبط مع حقيقة أن الجسم لا ينتج صبغة الميلانين) ،
- البقع الداكنة على البشرة الفاتحة (نقص التصبغ بسبب عدم كفاية إنتاج الميلانين)
- الأعراض الجسدية أو الخارجية: أمراض العين مثل الحول أو ضمور العصب البصري ،
- انحناء العمود الفقري (الجنف) ،
- الساقين المتصلبة (عندما يمشي رجل لا ينحني ركبتيه بسبب الحركة الصغيرة للمفاصل ، وبالتالي المقارنة مع مشية العرائس).
- الأعراض المرتبطة بالتطور العقلي والعاطفي:
- تأخر قوي في النمو العقلي ،
- سلوك عاطفي ، صاخب ، صعب بلا داع ،
- التصفيق المتكرر ،
- الصداقة التي تم التعبير عنها ، والتي تم إبرازها بابتسامة ثابتة على الوجه ،
- ضحك متكرر بلا سببه.
- الأعراض العصبية:
- هزة من الأطراف ،
- التنسيق غير الكافي للحركات مع فقدان التوازن ،
- انخفاض قوة العضلات ،
- مجموعة متنوعة من اضطرابات النوم ،
- نوبات هستيرية متكررة في مرحلة الطفولة ،
- ضعف الكلام (يبدأ الطفل في التكلُّم متأخرة ، ولديه مهارات اتصال ضعيفة وخطأ في الكلام) ،
- فرط النشاط على خلفية من زيادة استثارة ،
- صعوبات في التركيز والتدريب.
لكن هذه صورة عامة للمرض. في الواقع ، تعتمد الصورة السريرية لمتلازمة أنغلمان بشكل كبير على مرحلة تطور المرض ونوع الطفرة الصبغية التي سببت المرض. وهذا يعني أنه في المرضى المختلفين قد تختلف أعراض المرض بشكل كبير ، والتي لم تسمح لوقت طويل بعزل الأمراض من بين آخرين لديهم صورة سريرية مماثلة.
من بين العدد الكلي للأعراض يمكن تحديد هذه الخاصية التي تتميز بها جميع المرضى دون استثناء:
- الانحرافات الشديدة في النمو العقلي ،
- السلوك غير الكافي (ضحك بدون سبب ، زيادة الاستثارة ، ضعف التركيز ، حالة من النشوة) ،
- تخلف المهارات الحركية ،
- ضعف التنسيق للحركات ، ترنح المشي (تفاوت وتيرة ، يتأرجح من جانب إلى آخر ، وما إلى ذلك) ، هزة من الأطراف.
- انتهاك تطور الكلام مع غلبة وسائل الاتصال غير اللفظية.
من بين الأعراض التي تحدث في الغالبية العظمى من المرضى ، يمكننا تمييز ذلك:
- غير متناسب الرأس والجذع ، والناجمة عن التأخير في النمو البدني ،
- في كثير من المرضى يكون شكل الجمجمة بحيث يبقى حجم الدماغ أصغر من الأشخاص الأصحاء (صغر الرأس) ،
- نوبات الصرع في سن تصل إلى 3 سنوات مع انخفاض تدريجي في القوة والتردد في كبار السن ،
- تشوه مؤشرات EEG (التذبذبات والسعة العالية للموجات ذات التردد المنخفض).
هذه الأعراض تحدث في كثير من الأحيان ، ولكن في 20 ٪ من المرضى الذين يعانون من متلازمة انجلمان هم غائبون.
حتى أكثر نادرا يمكن تشخيص هذه المظاهر للمرض على النحو التالي:
- الحول أو طفيف الحول ،
- سيطرة ضعيفة على حركة اللسان ، ونتيجة لذلك المرضى في كثير من الأحيان التمسك لسانهم من دون سبب ،
- صعوبات في البلع والامتصاص ، خاصة عند الأطفال الصغار ،
- انتهاك الجلد وتصبغ العين ،
- رفع أو عازمة أثناء المشي ،
- giperrefleksiya،
- اضطرابات النوم ، وخاصة في مرحلة الطفولة ،
- اللعاب المتكرر ،
- عطش لا يمكن كبته ،
- حركات المضغ النشطة بشكل مفرط
- فرط الحساسية للحرارة ،
- رأس مسطح
- الفك السفلي المتقدم ،
- النخيل على نحو سلس.
تعاني نسبة كبيرة من المرضى من مشاكل التبول ، والتي يسيطرون عليها بشكل سيئ ، وانتهاك المهارات الحركية الدقيقة ، مما يخلق صعوبات في الخدمة الذاتية والتدريب ، زيادة الوزن. عمليا في جميع المرضى يبدأ سن البلوغ في وقت لاحق من أقرانهم الأصحاء.
الأطفال الذين يعانون من متلازمة angelman جيدون في فهم وفهم الكلام الشفهي ، لكنهم لا يرغبون في المشاركة في المحادثة ، مما يحد من خطبهم إلى عشرات الكلمات الضرورية في الحياة اليومية. ولكن في مرحلة البلوغ ، يبدو هؤلاء المرضى أصغر سناً من أقرانهم دون أمراض وراثية.
العديد من أعراض متلازمة انجلمان متقلبة ، وبالتالي فإن الصورة السريرية للمرض تتغير مع تقدم العمر. وتصبح نوبات الصرع ونوبات الصرع أكثر ندرة أو تختفي على الإطلاق ، ويصبح المريض أقل تهيجًا ، وينتقل إلى النوم.
المضاعفات والنتائج
متلازمة انجلمان هي مرض خطير ، لا يمكن علاجه عمليًا لعلم الأمراض الصبغية ، والذي يحرم المرضى من فرصة العيش حياة طبيعية. ما سيكون حياة الطفل مع SA ، يعتمد إلى حد كبير على نوع من الشذوذ الكروموسومي.
إن ازدواجية منطقة الكروموسومات في معظم الحالات لا تتوافق مع الحياة. وحتى لو لم يمت هؤلاء المرضى في سن الرضاعة والوصول إلى سن البلوغ ، فإنهم لا يستطيعون إنجاب الأطفال.
إن حذف أو غياب جزء من الجينات التي تحدث في متلازمة أنجلمان غالباً ما يشكل عقبة أمام تعلم الطفل المشي والتحدث. في مثل هؤلاء الأطفال ، يتم عرض التخلف العقلي في شكل أكثر حدة ، وغالبا ما تحدث هجمات الصرع ، وشدتها أقوى بكثير من المرضى الذين يعانون من تشوهات الكروموسومات الأخرى.
إذا لم يكن هناك سوى طفرة في جين واحد ، مع الاهتمام والنهج المناسبين ، يمكن تعليم الطفل أساسيات الخدمة الذاتية والتواصل والاتصال في الفريق ، على الرغم من أنه لا يزال متخلفًا في التنمية من أقرانه.
بالنسبة للأطفال الذين يعانون من متلازمة أنجيليمان ، والطبيعة الخيرة ، فإن الحب هو واهتمام الوالدين. فقط في هذه الحالة ، سيؤثر تدريب الطفل ، حتى الأطفال الصغار. بطبيعة الحال ، لن يتمكن المرضى في المدرسة العادية من الدراسة مع SA. إنهم بحاجة إلى دروس خاصة ، حيث يتم تعليم الأطفال أولاً تركيز انتباههم ، ومن ثم سيعطون تدريجياً أساسيات المعرفة المدرسية.
التشخيص متلازمة انجلمان
متلازمة انجيلمان هي علم الأمراض الخلقي للتنمية. ولكن بسبب بعض الظروف ، فإن تشخيصها في مرحلة الطفولة والطفولة المبكرة غير ممكن في أغلب الأحيان. السبب في ذلك هو عدم النوعية والأعراض الخفيفة عند الرضع والأطفال حتى سن 3 سنوات. كما أن انتشار المرض في بلدنا ليس كبيرًا لدرجة أن الأطباء تعلموا كيف يتعرفون عليه بينه.
متلازمة أنجلمان عند الرضع يمكن أن يعبر عن نفسه في شكل انخفاض قوة العضلات، والذي يتجلى في شكل مشاكل التغذية (ضعف مص ومنعكس البلع)، وصعوبات في التعلم لاحقا إلى المشي (هؤلاء الأطفال تبدأ في وقت لاحق من ذلك بكثير المشي). هذه الأعراض هي العلامات الأولى للانحراف في نمو الجنين ، والذي قد يكون مرتبطا مع شذوذ صبغوي. تأكيد هذا الافتراض يمكن فقط التحليل الجيني.
يتم إيلاء اهتمام خاص للأطفال الذين لديهم آباء وأمهات مختلفين من التشوهات الجينومية أو الصبغية. بعد كل شيء ، لا يمكن للمرض أن يتجلى في أي شكل من الأشكال ، وإذا تم الكشف عن المرض في الوقت المناسب ، من خلال البدء في العمل الجاد مع الطفل ، فمن الممكن تحقيق نجاح أكبر بكثير في التدريب ، وإبطاء تطور المرض.
إذا كان الوالدان لديهما تشوهات صبغية مختلفة ، فإن التحليل الجيني يتم حتى قبل ولادة الطفل ، لأن CA هي واحدة من الأمراض التي يمكن اكتشافها في حالة جنينية.
يمكن جمع المواد للبحث الجيني بطريقتين:
- الغازية (مع نسبة مئوية معينة من المخاطر ، لأنه مطلوب للدخول إلى الرحم من أجل إجراء اختبار للسائل الأمنيوسي) ،
- غير الغازية (تحليل الحمض النووي للطفل عن طريق دم الأم).
ثم يتم إجراء البحوث التالية:
- التهجين الفلوري في الموقع (طريقة FISH) - الربط بين مسبار الحمض النووي المسمى بصبغة خاصة للحمض النووي تحت الدراسة ، تليها الفحص المجهري.
- تحليل الطفرات في الجين ube3a وطبع الجينات ،
- تحليل ميثيل الحمض النووي بمساعدة الطرق الخاصة المستخدمة في علم الوراثة.
تعطي التحليلات الوراثية معلومات دقيقة للغاية في حالة التشوهات الصبغية ، لذا فإن أولياء الأمور في المستقبل يعرفون مسبقاً ما يجب أن يكونوا مستعدين له. ومع ذلك ، هناك استثناءات. في مجموعة معينة من المرضى ، في ظل وجود جميع الأعراض التي تشير إلى أعراض ، فإن نتائج التحاليل تبقى طبيعية. أي للكشف عن الأمراض ، من الممكن فقط مراقبة الطفل عن كثب منذ الطفولة المبكرة: كيف يأكل ، عندما يبدأ في المشي والتحدث ، وما إذا كانت الأرجل تنحني عند المشي ، وما إلى ذلك.
بالإضافة إلى FISH-طريقة من بين طرق أداة تشخيصية متلازمة أنجلمان يمكن أن يكون التصوير المقطعي المميز (CT أو MRI)، مما يساعد على تحديد وضع وحجم الدماغ، والكهربائي (EEG)، والتي تبين كيف أن كل جزء من أجزاء الدماغ تعمل.
عادة ما يتم تحديد التشخيص النهائي للأطباء في سن 3-7 سنوات ، عندما يكون المريض يعاني بالفعل من غالبية الأعراض وديناميات تطور المرض مرئية.
ما هي الاختبارات المطلوبة؟
تشخيص متباين
متلازمة انجلمان هي مرض وراثي لا يوجد به في الواقع مظاهر محددة. معظم الأعراض يمكن أن تشير إلى كل من CA والأمراض الجينية الأخرى.
يتم التشخيص التفريقي في متلازمة أنجلمان مع الأمراض التالية:
- متلازمة بيت هوبكنز (يتصف المرضى بالتخلف العقلي ، والشخص البهيج ، ويبتسمون ، ولديهم فم كبير وكبير ، ويلاحظ صغر الرأس). الفرق - هجمات فرط التنفس وتأخير التنفس في حالة اليقظة.
- متلازمة Kristiansona (مرضى المتخلفين عقليا مع التصرف البهيج ، غير قادر على الكلام ، وتتميز صغر الرأس ، وترنح ، والتشنجات ، وحركات لا إرادية للعضلات).
- متلازمة مواتا ويلسون (الأعراض: التخلف العقلي ، نوبات الصرع ، الذقن الحادة ، الفم المفتوح ، التعبير عن السعادة على الوجه ، صغر الرأس). الفرق هو المسافة الكبيرة بين العينين ، يتم شطف العيون إلى الداخل ، يتم تقريب طرف الأنف ، يتم إرجاع الأوعية إلى الوراء.
- متلازمة كابوكي (تتميز بدرجة خفيفة ومتوسطة من التخلف العقلي ، مشكلات الكلام والحركية ، ضعف العضلات ، نوبات الصرع ، صغر الرأس ، فترات كبيرة بين الحكة ، ضعف التنسيق). الفرق - الحاجبين على شكل قوس ، جزء جانبي مقلوب من الجفن السفلي ، عيون واسعة ، شقوق طويلة في العين مع رموش طويلة سميكة.
- متلازمة ريت (التمايز مع CA في النساء). الأعراض: تأخر تطور الكلام ، النوبات التشنجية ، صغر الرأس. الفرق - لا يوجد تعبير سعيد على الوجه ، هناك هجمات انقطاع النفس و apraxia ، التي تتقدم في نهاية المطاف.
- متلازمة جسمية متنحية tardatsii العقلية 38 (الأعراض: التخلف العقلي مع تأخير ملحوظ في تطوير المهارات الحركية والكلام، وضعف العضلات، والتغذية المشاكل في مهدها والاندفاع). الفرق هو اللون الأزرق من القزحية.
- متلازمة تكرار الجين MESR 2 (تمايز مع SA في الرجال). الأعراض: تخلف عقلي حاد ، ضعف عضلي من الطفولة ، مشاكل في الكلام أو نقصان ، مرض الصرع. الاختلافات - اعتلال عضلي تدريجي ، استمرار العدوى المتكررة.
- متلازمة Clifstra (أعراض: مشاكل النطق والتفكير ، ضعف العضلات ، اضطرابات النوم ، عدم الاهتمام ، الفم المفتوح قليلاً ، فرط النشاط ، التشنجات ، الرنح ، عدم التوازن). الاختلافات - الوجه المسطح ، الانف الانف القصير ، العيون العريضة ، الشفة السفلى المقلوبة الكبيرة ، هجمات العدوان.
- متلازمة سميث-ماغنيس (تتميز بالنوبات المرضية ومشاكل النوم واضطرابات النمو الذهنية والحركية). الاختلافات - وجه واسع ومسطحة ، جبين محدب.
- متلازمة كولينا دي فرايز (التخلف العقلي المعتدل والخفيف ، ضعف العضلات ، الهجمات المتشنجة ، الود). الاختلافات - وجه طويل مع جبهته العالية ، وأذنيه بارزة ، وعيون مائلة ، وحركة أكبر للمفاصل ، وأمراض القلب الخلقية.
- متلازمة فيلان - مكدرميد (الأعراض: التخلف العقلي ، ضعف الكلام أو عدمه). الاختلافات - الأيدي الكبيرة مع العضلات المتقدمة ، وضعف العضلات منذ الولادة ، ضعف التعرق.
متلازمة أنجلمان أعراض مشابهة يمكن أن "التباهي" وكيف يمكن لهذا النقص adenilsuktsinazy علم الأمراض، ومتلازمة راثي جسمي مقهور من التخلف العقلي 1، والكروموسوم 2q23.1 متلازمة الازدواجية والجينات haploinsufficiency FOXG1، STXBP1 أو MEF2C وغيرها.
مهمة الطبيب هي إجراء تشخيص دقيق ، وتمييز متلازمة انجلمان من الأمراض التي لها أعراض مشابهة ، و وصف علاج فعال يكون ذا صلة بالدرجة التي تم تشخيصها من تطور المرض.
من الاتصال؟
علاج او معاملة متلازمة انجلمان
تشير متلازمة انجلمان إلى فئة تلك الأمراض ، والبحث عن العلاج الفعال الذي ينخرط فيه الدواء حتى يومنا هذا. العلاج المسبب للمرض هو في مرحلة التطوير من مختلف الوسائل والوسائل ، والكثير منها لم يتم اختباره بعد في البشر. يجب أن يكون مقصورا على علاج أعراض للمساعدة بطريقة أو بأخرى تخفيف معاناة الأطفال والبالغين الذين يعانون من متلازمة دمية، الذين يعانون من نوبات الصرع، واللعاب، انخفاض ضغط الدم، واضطرابات النوم الأطباء حتى الآن.
لذا يمكن تقليل تواتر وقوة نوبات الصرع باستخدام عقار مضاد للاختلاج يتم اختياره بشكل صحيح. لكن الصعوبة الكاملة هي أن النوبات في المرضى الذين يعانون من AS تختلف عن نوبات الصرع المعتادة في أنها تتميز بأنواع عديدة من التشنجات ، مما يعني أنه سيكون من الممكن تخفيف الحالة عن طريق تناول العديد من الأدوية دفعة واحدة.
أشهر مضادات الاختلاج المستخدمة لعلاج متلازمة أنجلمان هي: حمض الفالبوريك ، توبيراميت ، لاموتريجين ، ليففيتيراسيتام ، كلونازيبام والمستحضرات التي تعتمد عليها. أقل شيوعا المخدرات على أساس karmazepina، الفينيتوين، الفينوباربيتال، إيثوسكسيميد، لأن بعضها قد يؤدي إلى تأثير متناقض، هو تعزيز وزيادة وتيرة نوبات الصرع. يحدث هذا إذا تم استخدام الدواء كجزء من وحيد.
لعلاج اللعاب ، تستخدم عادة طريقتين: الطبية (التحضيرات قمع تكوين اللعاب) والتشغيلية ، والتي تتكون في إعادة زرع القنوات اللعابية. ولكن في حالة CA ، تعتبر هذه الطرق غير فعالة ، ويظل السؤال مفتوحًا. الآباء والأمهات والذين يهتمون لمثل هؤلاء المرضى ، علينا أن نولي اهتماما خاصا لهذه اللحظة ، لأن المرضى أنفسهم عادة لا يتحكمون في اللعاب ، وبعضهم ببساطة لا يستطيعون الاعتناء بأنفسهم.
مشكلة أخرى هي قصر مدة النوم. في كثير من الأحيان الأطفال الذين يعانون من متلازمة انجممان لا ينام أكثر من 5 ساعات ، مما يؤثر سلبا على عمل الكائن كله. يتعب الأطفال المثيرون والنشطون ، ألعاب الحب والتواصل (حتى لو حاولوا أن يحصروا أنفسهم بطرق غير لفظية) ، بشكل ملحوظ لليوم. للحصول على قسط جيد من الراحة ، يحتاج الجسم إلى نوم كامل ، ولكن هذا مجرد مشكلة في ذلك.
على ما يبدو، لتحسين النوم لدى المرضى الذين منفعل ينبغي أن تكون كافية المخدرات مع الآثار المهدئة (الفينوثيازين ومضادات الذهان غير التقليدية)، يهدئ الجهاز العصبي. ولكن في حالة CA ، فإن استخدام هذه الأدوية محفوف بمظهر الآثار السلبية. لذلك يفضل الأطباء الأدوية المنومة لا تزال الخفيفة، مثل "الميلاتونين" (إعداد الهرموني الطبيعي على أساس هرمون النوم)، والتي تعطي المرضى ساعة قبل الذهاب إلى الفراش في 1 قرص، و "هيدرامين". وتيرة العلاج والجرعة التي يتم تعيينها من قبل الطبيب ، وهذا يتوقف على حالة وعمر المريض.
أحيانا يعاني مرضى متلازمة angelman من مشاكل في الهضم والبراز. لضبط كرسي فمن الممكن عن طريق الاستعدادات ملين (أفضل من phytogenesis).
ويمكنك معالجة المشكلة بشكل مختلف، كما فعل الأطباء الأميركيين، استنادا إلى بعض طرق العلاج من مرض التوحد، لأن الكثير من الأعراض المميزة للSA، هي أيضا من سمات التوحد (الاندفاع، وحركات لا إرادية، والإجراءات المتكررة، والعجز في الانتباه، ومشاكل في الاتصال، الخ ) .. وقد لوحظ أن إدارة سيكريتين الهرمون، وتطبيع الهضم وكرسي، ولها تأثير إيجابي على الاهتمام المرضى والأوكسيتوسين يساعد على تحسين قدرات الأطفال المعرفية والذاكرة، لتصحيح السلوك.
صحيح أن بعض الهرمونات لا غنى عنها هنا ، خاصة عندما يتعلق الأمر بالأطفال. تظهر متلازمة انجلمان العلاج السلوكي ، والعمل مع طبيب نفساني ومعالج الكلام (تدريس طرق غير اللفظية للتواصل ولغة الإشارة). وينبغي أن يستند تدريب هؤلاء الأطفال إلى برنامج فردي بمشاركة مدرسين مدربين تدريبا خاصا ، وأخصائي علم نفس وآباء. للأسف ، هذا غير ممكن في كل مكان ، والأسر تبقى وحدها مع مشكلتها.
نظرًا لأن العديد من المرضى الصغار المصابين بمرض التصلب العصبي المتعدد يعانون من ضعف العضلات ومشاكل المفاصل ، يتم توجيه الكثير من الاهتمام للعلاج الفيزيائي العلاجي. في معظم الأحيان ، يلجأ الأطباء إلى استخدام تطبيقات البارافين ، والكهرومغناطيسي ، والمغناطيسية.
سيساعد تدليك التنغيم النشط والتمارين الخاصة للعلاج الطبيعي الطفل المريض بعد وقت يقف بثقة على قدميه والمشي. مفيدة بشكل خاص في هذا الصدد aquagymnastics ، الذي يوصى به في كاليفورنيا في الماء البارد. يزيد من نبرة العضلات ويعلم الطفل أن يمتلك جسمه ، وينسق الحركات.
علاج مضاد للاختلاج
أخطر الأعراض في متلازمة أنجلمان هي النوبات ، مثل نوبات الصرع. لوحظ هذا المرض في 80 ٪ من المرضى ، مما يعني أن جميعهم يجب وصفهم بعلاج فعال مضاد للاختلاج.
يتم علاج نوبات الصرع بمساعدة الفيتامينات ومضادات الاختلاج. عندما متلازمة أنجلمان، يرافقه أعراض المتشنجة، وسوف تكون مفيدة الفيتامينات مجموعة B، وكذلك الفيتامينات C، D و E. ولكن فيتامين العلاج تعيين تلقاء نفسها في هذه الحالة أمر خطير جدا، لأن كمية غير المنضبط من الفيتامينات قد تقلل من فعالية العقاقير المضادة للصرع وإثارة جديدة، أكثر حدة وأطول أمدا الهجمات.
وينبغي أيضا اختيار الأدوية المضادة للاختلاج وتعيين جرعتها الفعالة من قبل طبيب متخصص. وقال انه تقرر أن يكون ما يكفي من دواء واحد أو أن المريض يكون وقتا طويلا لاتخاذ 2 أو أكثر من الأدوية.
بالنسبة لمعظم المرضى ، يصف الأطباء مستحضرات حمض الفالبرويك (حمض فالبوريك ، ديباكين ، كونفوليكس ، فالبارين ، إلخ) ، والتي تمنع النوبات ، وتحسن الحالة المزاجية والحالة الذهنية للمرضى.
حمض Valproic متاح في شكل أقراص ، شراب ومحاليل عن طريق الحقن. الدواء الأكثر شعبية هو المخدرات لفترات طويلة "Depakin" في أقراص وكحل للإعطاء عن طريق الوريد. يتم تحديد جرعة الدواء من قبل الطبيب على حدة اعتمادا على وزن وعمر وحالة المريض.
تناول الدواء أثناء الوجبات من 2 إلى 3 مرات في اليوم. متوسط الجرعة اليومية هو 20-30 ملغ لكل 1 كيلوغرام من وزن المريض ، والحد الأقصى هو 50 ملغ / كغ في اليوم الواحد.
موانع لاستخدام. لا يستخدم لانتهاكات الكبد والبنكرياس ، أهبة النزفية ، التهاب الكبد ، البورفيريا وفرط الحساسية للدواء.
من بين الآثار الجانبية يمكن أن يكون مرتبك من ناحية الارتجاف ، والهضم والبراز ، والتغيرات في وزن الجسم.
"Topiramate" هو أيضا الدواء المفضل في كاليفورنيا. وهي مصنوعة في شكل أقراص وتستخدم كجزء من وحيد ، وبالاقتران مع أدوية أخرى.
طريقة التطبيق والجرعة. تأخذ حبوب منع الحمل في الداخل دون النظر إلى تناول الطعام. المدخول اليومي الأولي للكبار هو 25-50 ملغ ، للأطفال 0.5-1 ملغم / كغم. كل أسبوع ، يتم زيادة الجرعة وفقا لوصفة الطبيب.
لا ينبغي أن يؤخذ الدواء أثناء الحمل والرضاعة ، وكذلك مع زيادة الحساسية لمكوناته. الدواء له العديد من الآثار الجانبية المختلفة.
المخدرات أن الطبيب يمكن أن يصف في متلازمة أنجلمان "Klomazepam"، "Rivotril" "اموتريجين"، "Seyzar"، "Lamictal"، "ليفي تيرا سيتام"، "Keppra"، "Epiterra" وآخرون.
العلاج البديل والمعالجة المثلية
يختلف الطب البديل ، مثل العقاقير المثلية ، بالتأكيد عن الأمان المقارن ، ولكن هنا يمكن اعتبار فعالية مثل هذا العلاج فيما يتعلق بمتلازمة انجلهولم مثيرة للجدل.
على الرغم من أن بعض العلاجات البديلة لا يزال من الممكن المساعدة. انها عن وقف نوبات الصرع. في هذا الصدد ، يمكن أن يكون العلاج بالأعشاب فعالة للغاية.
يتم توفير تأثير جيد من خلال رسوم طبية على أساس الفاوانيا وعرق السوس و duckweed (تؤخذ المكونات بكميات متساوية). يجب أن يتم طحن الأعشاب إلى دقيق. بعد أسبوعين من بداية حفل الاستقبال ، يمكنك ملاحظة انخفاض كبير في وتيرة الهجمات المتشنجة.
مفيد لتشنجات وديكوجن من الخزامى (1 ملعقة صغيرة لكوب من الماء المغلي). يتم غلي الصيغة لمدة 5 دقائق وأصرت لمدة نصف ساعة. تناول الدواء طوال الليل لمدة 14 يومًا.
يعتبر فعالا في حالات نوبات الصرع هو الماء (أو الكحول).
من الأدوية المثلية لمنع النوبات مع متلازمة أنجلمان يمكن استخدام الأدوية على أساس البابونج وmotherwort، وحامض hydrocyanicum، فضة nitricum، كاليوم bromatum، ال Arsenicum الألبوم. ولكن من الضروري أن نعتبر أن الجرعات الفعالة والآمنة من الاستعدادات في كل حالة محددة يمكن أن تعين الطبيب المعالج المثلي فقط.
الوقاية
وكما قد يكون القارئ قد فهم بالفعل ، لمنع تحول الجينات وغيرها من التشوهات الصبغية ، لا يزال الدواء خارج نطاق السلطة ، ومع ذلك ، لعلاج الموقف. هذا يمكن أن يحدث للجميع ، لأن الأطفال الذين يعانون من متلازمة أنجلمان يولدون أيضًا في أهل أصحاء ، وعلم الوراثة ، الذي هو في الوقت الحالي واحد من فروع الطب الأقل دراسته ، لا يمكن أن يفسر ذلك بعد.
الشيء الوحيد الذي يمكن القيام به هو تحمل المسؤولية المستقبلة لتخطيط الحمل ، ليتم تسجيلها وفحصها في الوقت المناسب. ولكن مرة أخرى ، فإن مثل هذا الإجراء لن يكون وقفا ، بل إدراكي ، مثل أي مسح. إلا أن أولياء الأمور الصغار سلفًا سيعرفون ما الذي يجب الاستعداد له ، وفي حالة الإجابة الإيجابية ، سيقررون ما إذا كانوا قادرين على تحمل المسؤولية مثل تربية طفل مريض.
توقعات
يعتمد تشخيص متلازمة أنغلمان على طبيعة الشذوذ الكروموسومي وتوقيت الكشف عنه. الجزء الأصعب بالنسبة لأولئك الأطفال الذين تحتوي على 15 كروموسومات تحتوي على جينات "مفقودة" (الحذف). احتمال المشي والتحدث في مثل هؤلاء المرضى هو صغير للغاية. الحالات المتبقية مع اتباع نهج الاهتمام والحب لطفلك قابلة للتصحيح.
مثل هؤلاء المرضى ، للأسف ، لا يمكن أن يصبحوا أعضاء كاملين في المجتمع ، على الرغم من حقيقة أنهم بعيدون كل البعد عن الغباء ، فهم يفهمون الكلام ومعناه. هنا مجرد مشاكل مع التواصل لديهم في الحياة. يمكن تعليم المرضى لغة الإشارة منذ الطفولة ، لكن لا يمكن إجبار المرء على التواصل مع الكلمات. يقتصر قاموس "الحديث" على الحد الأدنى من الكلمات المستخدمة في الحياة اليومية (5-15 كلمة).
أما بالنسبة لمتوسط العمر المتوقع والصحة العامة للمرضى الذين يعانون من متلازمة أنجلمان ، فإن الأرقام هنا تتقلب في المتوسط. في مرحلة البلوغ ، يعاني المرضى عمومًا من مشاكل صحية مثل الجنف والسمنة ، التي لا تهدد الحياة ، مع النهج الصحيح للعلاج.