خبير طبي في المقال

منشورات جديدة
متلازمة أنجلمان لدى الأطفال والبالغين
آخر مراجعة: 04.07.2025

تتم مراجعة جميع محتويات iLive طبياً أو التحقق من حقيقة الأمر لضمان أكبر قدر ممكن من الدقة الواقعية.
لدينا إرشادات صارمة من مصادرنا ونربط فقط بمواقع الوسائط ذات السمعة الطيبة ، ومؤسسات البحوث الأكاديمية ، وطبياً ، كلما أمكن ذلك استعراض الأقران الدراسات. لاحظ أن الأرقام الموجودة بين قوسين ([1] و [2] وما إلى ذلك) هي روابط قابلة للنقر على هذه الدراسات.
إذا كنت تشعر أن أيًا من المحتوى لدينا غير دقيق أو قديم. خلاف ذلك مشكوك فيه ، يرجى تحديده واضغط على Ctrl + Enter.

هناك عدد من الأمراض التي تبدو فيها عبارات مثل "اعتنِ بنفسك ولن تمرض" سخيفة، على أقل تقدير. هذه أمراضٌ تكون فيها بعض التشوهات العقلية والجسدية متأصلة في جسم الطفل حتى قبل الولادة، ولكن الوالدين غير مسؤولين عن ذلك. تحدث هذه الأمراض بسبب طفرات أو تشوهات في مجموعات الكروموسومات، وتُسمى كروموسوماتية أو وراثية. متلازمة أنجلمان، ومتلازمة داون، ومتلازمة باتو، ومتلازمة إدواردز، ومتلازمة تيرنر، ومتلازمة برادر-ويلي - هذه ليست سوى جزء من الأمراض الوراثية من قائمة جيدة إلى حد ما.
متلازمة الرجل السعيد
سنتحدث هذه المرة عن هذا المرض، الذي سُمي تيمنًا بطبيب الأطفال الإنجليزي هاري أنجلمان، الذي أثار هذه المشكلة لأول مرة عام ١٩٦٥، بعد أن صادف ثلاثة أطفال غير عاديين في عيادته في اليوم السابق، تجمعهم أعراض غريبة مشتركة. أطلق الطبيب على هؤلاء الأطفال اسم "أطفال الدمى" وكتب مقالًا عنهم، عُنون في البداية بـ"أطفال الدمى المتحركة". استوحى المقال وعنوانه من لوحة عُرضت في أحد متاحف فيرونا. تُصوّر اللوحة صبيًا ضاحكًا، وسُمّيت "صبي الدمية". دفع ارتباط الطفل المصوّر في اللوحة بالأطفال الثلاثة الذين صادفهم أنجلمان في عيادته طبيب الأطفال إلى دمج الأطفال في مجموعة واحدة نظرًا لمرضهم.
ليس من المستغرب أن الأطفال المذكورين في المقال لم يُلاحظهم أطباء آخرون. ففي النهاية، للوهلة الأولى، بدا أنهم مصابون بأمراض مختلفة تمامًا، إذ كانت الصورة السريرية العامة للمرض مختلفة جدًا في ثلاث حالات مختلفة. ربما كان علم الأمراض الكروموسومي "الجديد" سيثير اهتمام علماء آخرين، لكن علم الوراثة لم يكن قد تطور بما يكفي آنذاك لتأكيد فرضية الطبيب الإنجليزي. لذلك، بعد أن حظي المقال باهتمام معين، أُهمل لفترة طويلة.
يعود ذكر متلازمة أنجلمان، وهو الاسم الذي أُطلق على مقال طبيب الأطفال الإنجليزي ج. أنجلمان، إلى أوائل ثمانينيات القرن العشرين. ولم يُكتشف سبب ولادة عدد قليل من الأطفال بمثل هذه التشوهات إلا في عام ١٩٨٧، حيث يبدون من الخارج مبتسمين وسعداء باستمرار. في الواقع، هذا غير صحيح إطلاقًا، فالابتسامة مجرد تجهم يخفي وراءه تعاسة نفسية وألمًا يغمر الوالدين.
علم الأوبئة
وفقًا للإحصاءات، يمكن أن تتطور طفرة كروموسومية لدى الطفل، سواءً على خلفية طفرات مماثلة لدى الوالدين أو في غيابها. لا توجد طبيعة وراثية واضحة لمتلازمة أنجلمان (AS)، ولكن احتمالية الإصابة بالأمراض لدى الوالدين المصابين بطفرات كروموسومية عالية جدًا.
ومن المثير للاهتمام أيضًا أنه إذا كانت الأسرة لديها بالفعل طفل مصاب بمتلازمة أسبرجر، فهناك احتمال بنسبة واحد بالمائة لإنجاب طفل ثانٍ مصاب بنفس الاضطراب، حتى لو كان الوالدان بصحة جيدة.
لا توجد حتى الآن إحصائيات دقيقة حول عدد مرضى متلازمة أنجلمان. ولعل السبب هو تنوع الأعراض، التي قد تظهر بتركيبة معينة أو تختفي تمامًا لفترة طويلة. ويُفترض أن معدل انتشار المرض هو طفل واحد لكل 20,000 مولود جديد. إلا أن هذا الرقم تقريبي للغاية.
الأسباب متلازمة أنجلمان
متلازمة أنجلمان اسم طبي لمرض كروموسومي، لكنها ليست الوحيدة. يُطلق الناس على هذا المرض أسماءً متنوعة (قد تكون مسيئة للمرضى أنفسهم وذويهم)، لكن المرض يبقى مرضًا، مهما بدا مضحكًا ومهما كانت أسبابه.
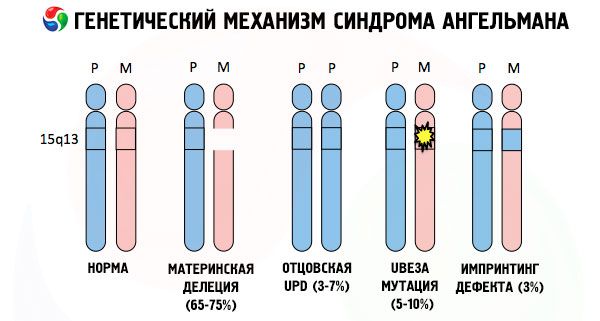
وأسباب تطور متلازمة أنجلمان، كما هو الحال مع العديد من الأمراض الوراثية الأخرى، هي في جميع الحالات اضطرابات في بنية أحد الكروموسومات أو مجموعة الكروموسومات ككل. ولكن في حالتنا، تكمن المشكلة برمتها في الكروموسوم 15، الموروث من الأم. أي أن الكروموسوم الأبوي في هذه الحالة خالٍ من أي انحرافات، بينما يتعرض الكروموسوم الأنثوي لبعض الطفرات.
وفقًا لنوع الخلل الكروموسومي، تُصنف متلازمة أنجلمان كطفرة كروموسومية. تُعتبر هذه الطفرات:
- الحذف (غياب جزء من الكروموسوم يحتوي على مجموعة معينة من الجينات؛ إذا كان أحد الجينات مفقودًا، فإننا نتحدث عن حذف مجهري)، وهو نتيجة لانقطاعين وإعادة توحيد واحد، عندما يتم فقدان جزء من الكروموسوم الأصلي.
- التضاعف (وجود قسم إضافي في الكروموسوم يكون نسخة من قسم موجود)، والذي يؤدي في أغلب الحالات إلى وفاة الشخص، وفي حالات أقل إلى العقم.
- الانقلاب (انعكاس أحد أقسام الكروموسوم بمقدار 180 درجة، أي في الاتجاه المعاكس، ومن ثم تقع الجينات فيه بالترتيب المعاكس)، عندما تتصل الأطراف المكسورة من الكروموسوم بترتيب مختلف عن الترتيب الأصلي.
- الإدخال (إذا كان جزء من المادة الوراثية في الكروموسوم خارج مكانه)،
- الانتقال (إذا كان جزء معين من الكروموسوم مرتبطًا بكروموسوم آخر؛ يمكن أن تكون هذه الطفرة متبادلة دون فقدان الأجزاء).
عند تلقي الطفل كروموسومًا متحورًا من أم غافلة، يُحكم عليه بالولادة بتشوهات خلقية. ولا يزال السبب الأكثر شيوعًا لمتلازمة أنجلمان هو حذف الكروموسوم الخامس عشر من الأم، عند فقدان جزء صغير منه. أما الطفرات الأقل شيوعًا في متلازمة "الدمية الضاحكة" فتشمل:
- النقل،
- ثنائي الصبغيات من الأب (إذا تلقى الطفل زوجًا من الكروموسومات من الأب، يكون الكروموسوم الأمومي غائبًا)،
- طفرة الجينات في الحمض النووي، والتي تعد المادة البنائية (الوراثية) الرئيسية وتعليمات استخدامها الصحيح (على وجه الخصوص، طفرة جين ube3a في الكروموسوم الأمومي).
يُعد وجود إحدى هذه الطفرات لدى الوالدين عامل خطر للإصابة بمتلازمة أنجلمان لدى الأطفال. ولكن ليس الطفرات الكروموسومية وحدها هي التي تُحفز ظهور المرض لدى الطفل، بل أيضًا الطفرات الجينومية (التي ترتبط بتغير كمي في مجموعات الكروموسومات، وهي أكثر شيوعًا من الطفرات الكروموسومية). ومن الطفرات الجينومية الشائعة التثلث الصبغي (إذا كانت مجموعة الكروموسومات لدى الشخص تحتوي على أكثر من 46 كروموسومًا).
لظهور مرضٍ ما لدى الطفل، ليس من الضروري إطلاقًا أن يكون لدى الوالدين خللٌ كروموسومي. ومع ذلك، هناك نسبةٌ معينةٌ من المرضى يكون مرضهم وراثيًا.
طريقة تطور المرض
لنتعمق أكثر في علم الأحياء، أو بالأحرى، علم الوراثة. تحتوي كل كائن حي بشري على 23 زوجًا من الكروموسومات. ينتقل أحد الكروموسومين من الأب إلى الطفل، والآخر من الأم. تختلف جميع أزواج الكروموسومات في الشكل والحجم، وتحمل معلومات معينة. وبالتالي، فإن الزوج الثالث والعشرون من الكروموسومات (الكروموسومان X وY) مسؤول عن تكوين الصفات الجنسية للطفل (XX للأنثى، وXY للذكر، بينما لا يرث الطفل الكروموسوم Y إلا من الأب).
في الحالة المثالية، يتلقى الطفل 46 كروموسومًا من والديه، تُشكل هذه الكروموسومات صفاته الوراثية، وتُحدد شخصيته مسبقًا. يُطلق على العدد الأكبر من الكروموسومات اسم "التثلث الصبغي"، ويُعتبر انحرافًا عن القاعدة. على سبيل المثال، يُسبب وجود الكروموسوم 47 في مجموعة الكروموسومات (النمط النووي، وتحديد النوع، والصفات الفردية) حدوث متلازمة داون.
إذا صُبغت الكروموسومات بصبغة خاصة، يُمكن رؤية خطوط بألوان مختلفة على طول كل منها تحت المجهر. يحتوي كل خط على عدد هائل من الجينات. جميع هذه الخطوط مُرقمة علميًا ولها موقع ثابت. يُعتبر غياب أي منها انحرافًا عن القاعدة. في متلازمة أنجلمان، غالبًا ما يُلاحظ غياب أجزاء من الكروموسوم الأمومي في الفترة q11-q13، الواقعة في الذراع الطويلة، والتي يبلغ عدد قواعد الحمض النووي فيها حوالي 4 ملايين قاعدة فقط.
يُعتبر المكوّن الرئيسي للكروموسوم جزيء DNA طويلًا للغاية، يحتوي على آلاف الجينات وعشرات ومئات الملايين من القواعد النيتروجينية. وهكذا، يحتوي الكروموسوم 15، المسؤول عن تطور متلازمة أنجلمان والعديد من الأمراض الأخرى، على 1200 جين وحوالي 100 مليون قاعدة. أيّ خلل في بنية جزيء DNA سيؤثر حتمًا على مظهر الطفل المستقبلي ونموه.
تُحوَّل المعلومات الوراثية الموجودة في الجينات إلى بروتين أو حمض نووي ريبوزي (RNA). تُسمى هذه العملية التعبير الجيني. بهذه الطريقة، تكتسب المعلومات الوراثية المُستقاة من الوالدين شكلاً ومضموناً، ويتجسد ذلك في وريثهم الفريد، ذكراً كان أو أنثى.
هناك عدد من الأمراض ذات النوع غير الكلاسيكي من الوراثة، بما في ذلك متلازمة أنجلمان، حيث تحمل الجينات المستلمة من الوالدين كجزء من الكروموسومات المزدوجة بصمة فريدة من نوعها للوالدين وتتجلى بطرق مختلفة.
لذا، تُعدّ متلازمة أنجلمان مثالاً بارزاً على البصمة الجينية، حيث يعتمد التعبير الجيني في جسم الطفل بشكل مباشر على الوالد الذي تلقّى منه الأليلات (أشكال مختلفة من جين واحد، مأخوذة من الأب والأم، وتقع في أجزاء متطابقة من الكروموسومات المزدوجة). أي أن الشذوذ في الكروموسوم الأمومي فقط هو الذي يؤدي إلى ظهور المتلازمة، بينما تُسبب الطفرات والاضطرابات البنيوية في الكروموسوم الأبوي أمراضاً مختلفة تماماً.
في هذه الحالة المرضية، يوجد نقص في جينات معينة في الكروموسوم الأمومي، أو نقص/انخفاض في نشاط جينات فردية (في الغالبية العظمى من الحالات، جين ube3a، الذي يشارك في استقلاب اليوبيكويتين، وهو بروتين ينظم تحلل البروتينات الأخرى). ونتيجة لذلك، يُشخَّص الطفل بتشوهات في النمو العقلي والجسدي.
الأعراض متلازمة أنجلمان
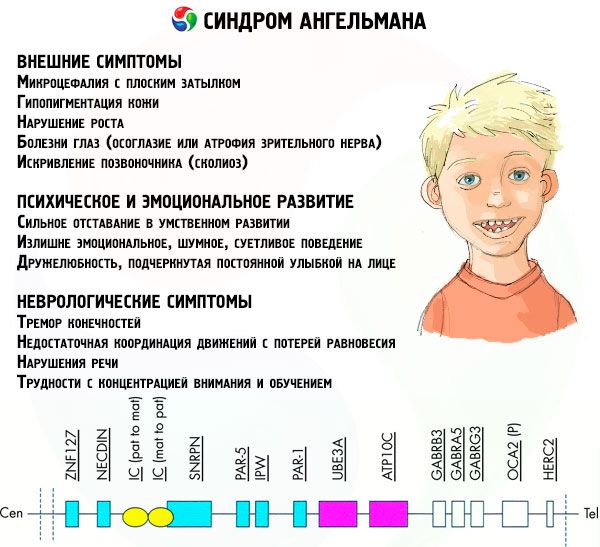
تؤثر أعراض متلازمة أنجلمان على جوانب مختلفة من حياة الطفل ونموه: الجسدية، والعصبية، والعقلية. وبناءً على ذلك، يمكن تحديد ثلاث مجموعات من الأعراض التي تشير إلى تطور هذه الحالة المرضية.
- الأعراض الخارجية أو الجسدية:
- رأس صغير بشكل غير متناسب مقارنة بالجسم والأطراف، والتي هي ذات حجم طبيعي،
- فم واسع جدًا،
- هناك دائمًا ابتسامة على الوجه (بفم مفتوح)،
- أسنان متفرقة،
- الشفة العليا الضيقة،
- لسان عريض بارز بشكل متكرر،
- بروز الفك السفلي،
- ذقن مدببة،
- بشرة فاتحة جدًا، وغالبًا ما يكون هناك شعر (المهق، المرتبط بحقيقة أن الجسم لا ينتج صبغة الميلانين)،
- بقع داكنة على البشرة الفاتحة (نقص التصبغ بسبب عدم كفاية إنتاج الميلانين)
- الأعراض الجسدية أو الخارجية: أمراض العيون مثل الحول أو ضمور العصب البصري،
- انحناء العمود الفقري (الجنف)،
- تيبس الساقين (عند المشي لا يقوم الشخص بثني ساقيه عند الركبتين بسبب قلة حركة المفاصل، ومن هنا جاءت المقارنة مع مشية الدمية).
- الأعراض المتعلقة بالتطور العقلي والعاطفي:
- التخلف العقلي الشديد،
- سلوك عاطفي مفرط، صاخب، متطلب،
- التصفيق المتكرر للأيدي،
- أعرب عن ودّه، وأكد ذلك بابتسامة دائمة على وجهه،
- الضحك المتكرر بدون سبب.
- الأعراض العصبية:
- ارتعاش الأطراف،
- عدم كفاية تنسيق الحركات مع فقدان التوازن،
- انخفاض قوة العضلات،
- اضطرابات النوم المختلفة،
- نوبات الهستيريا المتكررة في مرحلة الطفولة،
- اضطرابات الكلام (يبدأ الطفل في التحدث متأخرًا، ولديه مهارات تواصل ضعيفة وكلام غير واضح)،
- فرط النشاط على خلفية زيادة الإثارة،
- صعوبات في التركيز والتعلم.
لكن هذه صورة عامة للمرض. في الواقع، تعتمد الصورة السريرية لمتلازمة أنجلمان بشكل كبير على مرحلة تطور المرض ونوع الطفرة الكروموسومية المسببة له. هذا يعني أن أعراض المرض قد تختلف اختلافًا كبيرًا بين المرضى، مما حال دون تمييزه عن غيره من الأمراض ذات الصورة السريرية المشابهة لفترة طويلة.
ومن بين العدد الإجمالي للأعراض، يمكننا تسليط الضوء على تلك التي تميز جميع المرضى دون استثناء:
- التخلف العقلي الشديد،
- السلوك غير المناسب (الضحك غير المعقول، زيادة الإثارة، ضعف التركيز، حالة من النشوة)،
- ضعف تنمية المهارات الحركية،
- ضعف تنسيق الحركات، خلل في المشي (سرعة غير متساوية، التأرجح من جانب إلى آخر، وما إلى ذلك)، ارتعاش الأطراف.
- اضطراب تطور الكلام مع غلبة وسائل التواصل غير اللفظية.
ومن بين الأعراض التي تظهر على الغالبية العظمى من المرضى يمكن تمييز ما يلي:
- عدم التناسب بين الرأس والجسم بسبب التأخر في النمو البدني،
- في العديد من المرضى يكون شكل الجمجمة بحيث يظل حجم الدماغ أصغر من حجمه في الأشخاص الأصحاء (صغر الرأس)،
- النوبات الصرعية قبل سن 3 سنوات مع انخفاض تدريجي في القوة والتكرار في سن أكبر،
- تشويه معلمات تخطيط كهربية الدماغ (التقلبات والسعة العالية للموجات منخفضة التردد).
تعتبر هذه الأعراض شائعة جدًا، ومع ذلك، لا يعاني 20% من مرضى متلازمة أنجلمان من هذه الأعراض.
وفي حالات أقل شيوعاً، من الممكن تشخيص مظاهر المرض مثل:
- الحول الشديد أو الخفيف،
- ضعف التحكم في حركة اللسان، مما يؤدي في كثير من الأحيان إلى إخراج المرضى لسانهم دون سبب،
- صعوبات في البلع والامتصاص، وخاصة عند الأطفال الصغار،
- اضطراب تصبغ الجلد والعين،
- رفع الذراعين أو ثنيهما أثناء المشي،
- فرط المنعكسات،
- اضطرابات النوم، وخاصة في مرحلة الطفولة،
- كثرة إفراز اللعاب،
- عطش لا يشبع،
- حركات المضغ المفرطة النشاط،
- فرط الحساسية للحرارة،
- الجزء الخلفي المسطح من الرأس،
- بروز الفك السفلي،
- راحة اليد الناعمة.
يعاني عدد كبير من المرضى من مشاكل في التبول، يصعب عليهم التحكم فيه، ويعانون من ضعف في المهارات الحركية الدقيقة، مما يُصعّب عليهم العناية بأنفسهم والتعلم، بالإضافة إلى زيادة الوزن. ويتأخر سن البلوغ لدى جميع المرضى تقريبًا مقارنةً بأقرانهم الأصحاء.
يُدرك الأطفال المصابون بمتلازمة أنجلمان الكلام الشفهي جيدًا ويفهمونه، لكنهم لا يرغبون في المشاركة في أي محادثة، ويقتصر كلامهم على بضع عشرات من الكلمات الضرورية للحياة اليومية. ومع ذلك، في مرحلة البلوغ، يبدو هؤلاء المرضى أصغر سنًا من أقرانهم الذين لا يعانون من أمراض وراثية.
العديد من أعراض متلازمة أنجلمان غير ثابتة، لذا تتغير الصورة السريرية للمرض بشكل ملحوظ مع التقدم في السن. تقل وتيرة التشنجات والنوبات الصرعية أو تختفي تمامًا، ويصبح المريض أقل انفعالًا، ويتحسن نومه.
المضاعفات والنتائج
متلازمة أنجلمان هي مرض كروموسومي خطير، يكاد يكون من المستحيل علاجه حاليًا، ويحرم المرضى من فرصة عيش حياة طبيعية. وتعتمد حياة الطفل المصاب بمتلازمة أسبرجر بشكل كبير على نوع الخلل الكروموسومي.
في معظم الحالات، لا يتوافق تضاعف جزء من الكروموسوم مع الحياة. وحتى لو لم يمت هؤلاء المرضى في طفولتهم ولم يصلوا إلى سن البلوغ، فلن تكون لديهم فرصة للإنجاب.
يُعدّ حذف أو غياب جزء من الجينات، وهو الأكثر شيوعًا في متلازمة أنجلمان، عائقًا أمام تعلم الطفل المشي والكلام. يعاني هؤلاء الأطفال من شكل أكثر حدة من التخلف العقلي، كما تزداد نوبات الصرع لديهم، وتكون شدتها أكبر بكثير مقارنةً بالمرضى الذين يعانون من تشوهات كروموسومية أخرى.
إذا كان هناك طفرة في جين واحد فقط، فمن خلال الاهتمام والنهج المناسبين يمكن تعليم الطفل أساسيات العناية الذاتية والتواصل والتفاعل في المجموعة، على الرغم من أنه سيظل متأخرًا عن أقرانه في التطور.
بالنسبة للأطفال المصابين بمتلازمة أنجلمان، وهم طيبو القلب بطبيعتهم، فإن أهم شيء هو حب واهتمام والديهم. في هذه الحالة فقط، سيُثمر تعليم الطفل، حتى لو كان صغيرًا. بالطبع، لن يتمكن مرضى أسبرجر من الدراسة في المدارس العادية. إنهم بحاجة إلى فصول خاصة، حيث يُعلّمون الأطفال التركيز أولًا، ثم يُزوّدون تدريجيًا بأساسيات المعرفة المدرسية.
التشخيص متلازمة أنجلمان
متلازمة أنجلمان هي عيب خلقي في النمو. ولكن نظرًا لظروف معينة، غالبًا ما يستحيل تشخيصها في مرحلة الرضاعة والطفولة المبكرة. ويعود ذلك إلى عدم تحديد أعراضها وضعف ظهورها لدى الرضع والأطفال دون سن الثالثة. كما أن انتشار المرض في بلدنا ليس كبيرًا لدرجة أن الأطباء تعلموا تمييزه بين أقرانه.
قد تظهر متلازمة أنجلمان لدى الرضع على شكل ضعف في قوة العضلات، والذي يتجلى في مشاكل في التغذية (ضعف منعكس المص والبلع)، وصعوبات لاحقة في تعلم المشي (يبدأ هؤلاء الأطفال بالمشي في سن متأخرة جدًا). هذه الأعراض هي أولى علامات خلل في النمو لدى الطفل، والذي قد يكون مرتبطًا بخلل في الكروموسومات. ولا يمكن تأكيد هذا الافتراض إلا بالتحليل الجيني.
يُولى اهتمام خاص للأطفال الذين يُعاني آباؤهم من اضطرابات جينية أو كروموسومية مختلفة. ففي النهاية، قد لا يظهر المرض في البداية، وإذا كُشف عن المرض في الوقت المناسب، ومن خلال البدء في العمل المكثف مع الطفل، يُمكن تحقيق نجاح أكبر بكثير في التعلم، مما يُبطئ من تطور المرض.
إذا كان لدى الوالدين تشوهات كروموسومية مختلفة، يتم إجراء التحليل الجيني حتى قبل ولادة الطفل، لأن SA هو أحد الأمراض التي يمكن اكتشافها في المرحلة الجنينية.
يمكن جمع المواد اللازمة للبحث الجيني بطريقتين:
- - التوغلية (مع نسبة معينة من المخاطر، حيث أنه من الضروري اختراق الرحم من أجل أخذ عينة من السائل الأمنيوسي)،
- غير جراحي (تحليل الحمض النووي للطفل من دم الأم).
ويتم بعد ذلك إجراء الدراسات التالية:
- التهجين الموضعي الفلوري (طريقة FISH) - ربط مسبار DNA المسمى بصبغة خاصة بالحمض النووي الذي تتم دراسته، يليه الفحص تحت المجهر.
- تحليل الطفرات في جين ube3a والجينات المطبوعة،
- تحليل مثيلة الحمض النووي باستخدام طرق خاصة تستخدم في علم الوراثة.
تُقدم الاختبارات الجينية معلومات دقيقة إلى حد ما في حالة التشوهات الكروموسومية، مما يعني أن الوالدين المستقبليين يعرفون مسبقًا ما يجب عليهم الاستعداد له. ومع ذلك، هناك استثناءات. ففي مجموعة معينة من المرضى، ومع وجود جميع الأعراض التي تُشير إلى وجود مرض، تبقى نتائج الاختبار طبيعية. أي أنه لا يُمكن تحديد المرض إلا من خلال مراقبة الطفل بدقة منذ الطفولة المبكرة: طريقة تناوله للطعام، ومتى بدأ المشي والكلام، وما إذا كان يثني ساقيه أثناء المشي، وما إلى ذلك.
بالإضافة إلى طريقة FISH، من بين الطرق التشخيصية الآلية لمتلازمة أنجلمان، يمكن تمييز التصوير المقطعي (CT أو MRI)، الذي يساعد في تحديد حالة وحجم الدماغ، وتخطيط كهربية الدماغ (EEG)، الذي يوضح كيفية عمل أجزاء الدماغ الفردية.
يقوم الأطباء عادة بإجراء التشخيص النهائي في سن 3-7 سنوات، عندما يكون لدى المريض بالفعل معظم الأعراض وتكون ديناميكيات تطور المرض مرئية.
ما هي الاختبارات المطلوبة؟
تشخيص متباين
متلازمة أنجلمان هي مرض وراثي لا تظهر عليه أي أعراض محددة تقريبًا. تشير معظم الأعراض إلى كلٍّ من التهاب الفقار اللاصق وأمراض وراثية أخرى.
يتم إجراء التشخيص التفريقي لمتلازمة أنجلمان مع الأمراض التالية:
- متلازمة بيت هوبكنز (يتميز المرضى بتخلف عقلي، وشخصية مرحة، وابتسامة، وفم كبير وواسع، وصغر الرأس). الفرق هو نوبات فرط التنفس وحبس النفس أثناء اليقظة.
- متلازمة كريستيانسون (المرضى هم أشخاص متخلفون عقليًا يتمتعون بمزاج مرح، غير قادرين على الكلام، يتميزون بصغر الرأس، والترنح، والتشنجات، وحركات العضلات اللاإرادية).
- متلازمة موات ويلسون (أعراضها: تخلف عقلي، نوبات صرع، ذقن مدبب، فم مفتوح، تعبير وجه سعيد، صغر الرأس). المميزات: تباعد كبير بين العينين، انحناء العينين إلى الداخل، استدارة طرف الأنف، انعطاف صيوان الأذن إلى الخلف.
- متلازمة كابوكي (تتميز بتخلف عقلي خفيف إلى متوسط، ومشاكل في الكلام والحركة، وضعف عضلي، ونوبات صرع، وصغر الرأس، وحكة متقطعة، وضعف في التنسيق). تتميز بحاجبين مقوسين، وجزء جانبي مقلوب من الجفن السفلي، وعيون متباعدة، وشقوق جفنية طويلة مع رموش طويلة وسميكة.
- متلازمة ريت (التفريق بينها وبين متلازمة أسبرجر لدى النساء). الأعراض: تأخر في تطور الكلام، نوبات صرع، صغر الرأس. الفرق هو غياب تعبيرات السعادة على الوجه، ونوبات انقطاع النفس وفقدان القدرة على الحركة، والتي تتطور مع مرور الوقت.
- متلازمة التخلف العقلي الجسدي المتنحي 38 (الأعراض: تخلف عقلي ملحوظ مع تأخر في المهارات الحركية والكلام، وضعف عضلي، وصعوبات في التغذية في مرحلة الطفولة، واندفاعية). السمة المميزة هي اللون الأزرق للقزحية.
- متلازمة تضاعف جين MECP 2 (التمايز عن SA لدى الذكور). الأعراض: تخلف عقلي شديد، ضعف عضلي منذ الطفولة، صرع أو صعوبة في الكلام. العلامات المميزة: اعتلال عضلي تقدمي، التهابات متكررة.
- متلازمة كليفسترا (أعراضها: صعوبات في الكلام والتفكير، ضعف عضلي، اضطرابات في النوم، قلة انتباه، فم مفتوح، فرط نشاط، نوبات صرع، ترنح، اضطرابات في التوازن). السمات المميزة: وجه مسطح، أنف قصير أفطس، عينان متباعدتان، شفة سفلية كبيرة مقلوبة، نوبات غضب.
- متلازمة سميث-ماجينيس (تتميز بنوبات صرع، ومشاكل في النوم، واضطرابات في النمو العقلي والحركي). من السمات المميزة وجه عريض ومسطح وجبهة بارزة.
- متلازمة كولين دي فريس (تخلف عقلي خفيف إلى متوسط، ضعف عضلي، نوبات صرع، ود). السمات المميزة: وجه طويل بجبهة مرتفعة، آذان بارزة، عيون مائلة، حركة مفاصل عالية، عيوب خلقية في القلب.
- متلازمة فيلان-ماكديرميد (أعراضها: تخلف عقلي، اضطرابات في الكلام، أو فقدان القدرة على الكلام). السمات المميزة: أيادٍ كبيرة وعضلات متطورة، ضعف عضلي منذ الولادة، تعرق خفيف.
يمكن لأمراض مثل نقص سكسينات الأدينيل، ومتلازمة التخلف العقلي المتنحية الجسدية 1، ومتلازمة تكرار الكروموسوم 2q23.1، ومتلازمات نقص صبغي الجينات FOXG1 أو STXBP1 أو MEF2C وبعض الأمراض الأخرى أن "تتباهى" بأعراض مشابهة لمتلازمة أنجلمان.
مهمة الطبيب هي إجراء تشخيص دقيق، وتمييز متلازمة أنجلمان عن الأمراض ذات الأعراض المماثلة، ووصف العلاج الفعال المناسب للمرحلة التي تم تشخيص المرض فيها.
من الاتصال؟
علاج او معاملة متلازمة أنجلمان
متلازمة أنجلمان من الأمراض التي لا يزال الطب يبحث عن علاج فعال لها. ولا يزال العلاج المسبب للمرض في مرحلة التطوير، باستخدام أساليب ووسائل مختلفة، لم يُختبر الكثير منها على البشر بعد. وهذا يعني أن الأطباء يضطرون حاليًا إلى الاقتصار على العلاج العرضي، الذي يُساعد بطريقة ما على تخفيف الوضع المزعج للأطفال والبالغين المصابين بمتلازمة الماريونيت، والذين يعانون من نوبات صرع، وسيلان اللعاب، وانخفاض ضغط الدم، واضطرابات النوم.
وبالتالي، يُمكن تقليل وتيرة وشدة النوبات الصرعية باستخدام دواء مضاد للاختلاج مُختار بعناية. لكن تكمن الصعوبة في أن نوبات الصرع لدى مرضى الصرع الصرعي تختلف عن نوبات الصرع العادية من حيث أنها تتميز بأنواع متعددة من النوبات، مما يعني إمكانية تخفيف الحالة بتناول عدة أدوية في آن واحد.
أشهر مضادات الاختلاج المستخدمة لعلاج متلازمة أنجلمان هي: حمض الفالبرويك، توبيراميت، لاموتريجين، ليفيتيراسيتام، كلونازيبام، والأدوية المشتقة منها. أما الأدوية المشتقة من كارمازيبين، فينيتوين، فينوباربيتال، إيثوسكسيميد، فهي أقل استخدامًا، إذ قد يُسبب بعضها تأثيرًا متناقضًا يتمثل في زيادة وتيرة نوبات الصرع. ويحدث هذا عند استخدام الدواء كجزء من العلاج الأحادي.
لعلاج سيلان اللعاب، تُستخدم عادةً طريقتان: دوائيتان (أدوية تُثبّط إنتاج اللعاب) وجراحة، تتضمن إعادة زرع القنوات اللعابية. لكن في حالة التهاب الجيوب الأنفية، تُعتبر هذه الطرق غير فعّالة، ويبقى الأمر مفتوحًا. يجب على الآباء ومن يعتنون بهؤلاء المرضى إيلاء اهتمام خاص لهذه المسألة، لأن المرضى أنفسهم عادةً ما لا يستطيعون التحكم في سيلان اللعاب، وبعضهم ببساطة غير قادر على العناية بنفسه.
هناك مشكلة أخرى تتمثل في قلة مدة النوم. فغالبًا ما لا ينام الأطفال المصابون بمتلازمة أنجلمان أكثر من 5 ساعات، مما يؤثر سلبًا على وظائف الجسم ككل. فالأطفال سريعو الانفعال والنشيطون الذين يحبون الألعاب والتواصل (حتى لو حاولوا الاكتفاء بالأساليب غير اللفظية) يشعرون بتعب ملحوظ خلال النهار. وللحصول على قسط كافٍ من الراحة، يحتاج الجسم إلى نوم عميق وكامل، ولكن هذه هي المشكلة تحديدًا.
يبدو أن الأدوية المهدئة (الفينوثيازينات ومضادات الذهان غير التقليدية) التي تُهدئ الجهاز العصبي كافية لتحسين النوم لدى المرضى سريعي الانفعال. ولكن في حالة التهاب الفقار اللاصق، فإن استخدام هذه الأدوية محفوفٌ بآثار جانبية. لذلك، يُفضل الأطباء استخدام حبوب منومة خفيفة، مثل الميلاتونين (دواء هرموني طبيعي مُشتق من هرمون النوم)، والذي يُعطى للمرضى قبل ساعة من النوم بجرعة قرص واحد، وديفينهيدرامين. ويُحدد الطبيب وتيرة الإعطاء وجرعته بناءً على حالة المريض وعمره.
أحيانًا يعاني مرضى متلازمة أنجلمان من مشاكل في الهضم والبراز. يمكنك تحسين عملية الإخراج باستخدام الملينات (ويفضل أن تكون عشبية).
أو يُمكن التعامل مع المشكلة بطريقة مختلفة، كما فعل الأطباء الأمريكيون، بالاعتماد على بعض أساليب علاج التوحد، لأن العديد من الأعراض المميزة لمتلازمة أسبرجر هي أيضًا من سمات التوحد (الاندفاعية، والحركات اللاإرادية، والتكرار، ونقص الانتباه، ومشاكل التواصل، إلخ). وقد لوحظ أن إعطاء هرمون سيكريتين، الذي يُحسّن عملية الهضم والبراز، يُؤثر إيجابًا على انتباه المرضى، بينما يُساعد هرمون الأوكسيتوسين على تحسين القدرات الإدراكية والذاكرة لدى الطفل، وتصحيح سلوكه.
صحيح أن الهرمونات وحدها لا تكفي، خاصةً لدى الأطفال. في حالة متلازمة أنجلمان، يُنصح بالعلاج السلوكي، والعمل مع طبيب نفسي ومعالج نطق (تعليم أساليب التواصل غير اللفظي ولغة الإشارة). ينبغي أن يعتمد تعليم هؤلاء الأطفال على برنامج فردي بمشاركة معلمين مُدرَّبين تدريبًا خاصًا، وطبيب نفسي، وأولياء أمور. للأسف، هذا غير مُتاح في كل مكان، وتُترك العائلات وحدها مع مشكلتها.
نظراً لأن العديد من مرضى AS الشباب يعانون من ضعف في العضلات ومشاكل في المفاصل، يُولى اهتمام كبير للعلاج الطبيعي. وغالباً ما يلجأ الأطباء إلى استخدام دهانات البارافين، والرحلان الكهربائي، والعلاج المغناطيسي.
سيساعد التدليك المنشط والتمارين البدنية العلاجية الخاصة الطفل المريض على الوقوف والمشي بثقة بعد فترة. يُعدّ الجمباز المائي مفيدًا بشكل خاص في هذا الصدد، ويُنصح به في الماء البارد. فهو يزيد من قوة العضلات ويُعلّم الطفل التحكم في جسمه وتنسيق حركاته.
العلاج المضاد للاختلاج
أخطر أعراض متلازمة أنجلمان هي نوبات تشبه نوبات الصرع. يُلاحظ هذا العرض لدى 80% من المرضى، مما يعني أن جميعهم بحاجة إلى علاج فعال مضاد للاختلاج.
يُعالَجُ النوبات الصرعية باستخدام الفيتامينات ومضادات الاختلاج. في حالة متلازمة أنجلمان، المصحوبة بمتلازمة تشنجية، تُفيد فيتامينات المجموعة ب، بالإضافة إلى فيتامينات ج، د، وهـ. لكن وصف العلاج بالفيتامينات بشكل فردي في هذه الحالة أمرٌ خطيرٌ للغاية، لأن تناول الفيتامينات دون إشراف طبي قد يُقلل من فعالية مضادات الصرع ويُسبب نوبات جديدة أشد وطأةً وأطول أمدًا.
ينبغي أن يقوم طبيب مختص باختيار الأدوية المضادة للصرع ووصف جرعاتها الفعالة. وهو الذي يقرر ما إذا كان دواء واحد يكفي، أم سيحتاج المريض إلى تناول دواءين أو أكثر لفترة طويلة.
بالنسبة لمعظم المرضى، يصف الأطباء أدوية حمض الفالبرويك (حمض الفالبرويك، ديباكين، كونفيوليكس، فالبارين، وغيرها)، والتي تمنع النوبات وتحسن الحالة المزاجية والنفسية للمرضى.
يتوفر حمض الفالبرويك على شكل أقراص وشراب ومحاليل حقن. الدواء الأكثر شيوعًا هو دواء "ديباكين" ممتد المفعول، والذي يتوفر على شكل أقراص ومحلول وريدي. يُحدد الطبيب جرعة الدواء بناءً على وزن المريض وعمره وحالته الصحية.
يُؤخذ الدواء مع الوجبات مرتين إلى ثلاث مرات يوميًا. الجرعة اليومية المتوسطة هي ٢٠-٣٠ ملغ لكل كيلوغرام من وزن المريض، والحد الأقصى ٥٠ ملغ/كغ يوميًا.
موانع الاستعمال: لا يُستخدم في حالات اختلال وظائف الكبد والبنكرياس، أو فرط النزيف، أو التهاب الكبد، أو البورفيريا، أو فرط الحساسية للدواء.
وتشمل الآثار الجانبية ارتعاش اليد، واضطرابات الجهاز الهضمي والبراز، وتغيرات في وزن الجسم.
يُعد "توبيراميت" أيضًا دواءً مفضلًا لعلاج التهاب السحايا. يُنتج على شكل أقراص، ويُستخدم كجزء من العلاج الأحادي وبالاشتراك مع أدوية أخرى.
طريقة الاستخدام والجرعة. تناول الأقراص فمويًا بغض النظر عن تناول الطعام. الجرعة اليومية الأولية للبالغين هي ٢٥-٥٠ ملغ، وللأطفال ٠.٥-١ ملغ/كغ. تُزاد الجرعة أسبوعيًا وفقًا لتعليمات الطبيب.
لا يُنصح بتناول هذا الدواء أثناء الحمل والرضاعة، وكذلك في حالة فرط الحساسية لمكوناته. للدواء آثار جانبية عديدة ومتنوعة.
الأدوية التي قد يصفها الطبيب لمتلازمة أنجلمان: كلومازيبام، ريفوتريل، لاموتريجين، سيزار، لاميكتال، ليفاتيراسيتام، كيبرا، إيبيترا، إلخ.
 [ 16 ]، [ 17 ]، [ 18 ]، [ 19 ]
[ 16 ]، [ 17 ]، [ 18 ]، [ 19 ]
الطب التقليدي والمعالجة المثلية
وبطبيعة الحال، فإن الطب التقليدي، مثل المستحضرات المثلية، آمن نسبيًا، ولكن فعالية مثل هذا العلاج لمتلازمة أنجلمان يمكن اعتبارها مثيرة للجدل.
مع أن العلاج الشعبي قد يُساعد في بعض الأمور، إلا أننا نتحدث هنا عن إيقاف نوبات الصرع. وفي هذا الصدد، يُمكن أن يكون العلاج بالأعشاب فعالاً للغاية.
تُحقق مجموعة طبية مكونة من الفاوانيا وعرق السوس والطحالب المائية (تُؤخذ المكونات بكميات متساوية) تأثيرًا جيدًا. يجب طحن الأعشاب حتى تصبح دقيقًا. بعد أسبوعين من بدء تناولها، ستلاحظ انخفاضًا ملحوظًا في وتيرة النوبات.
مغلي اللافندر (ملعقة صغيرة لكل كوب من الماء المغلي) مفيد أيضًا للتقلصات. يُغلى المزيج لمدة 5 دقائق ويُنقع لمدة نصف ساعة. يُؤخذ الدواء ليلًا لمدة 14 يومًا.
يعتبر التسريب المائي (أو الكحولي) من عشبة الأم فعالاً في علاج النوبات الصرعية.
من بين المستحضرات المثلية للوقاية من النوبات في متلازمة أنجلمان، يُمكن استخدام أدوية مُصنّعة من البابونج وعشبة الأم، وحمض الهيدروسيانيك، والأرجنتوم النيتريك، وبروماتوم الكالسيوم، والزرنيخ الألبوم. ولكن يجب الأخذ في الاعتبار أن طبيب المعالجة المثلية وحده هو من يستطيع وصف جرعات فعالة وآمنة من الأدوية في كل حالة على حدة.
الوقاية
كما فهم القارئ على الأرجح، لا يزال الطب غير قادر على منع الطفرات الجينية وغيرها من التشوهات الكروموسومية، أو تصحيحها. هذا قد يحدث لأي شخص، لأن الأطفال المصابين بمتلازمة أنجلمان يولدون لوالدين سليمين، وعلم الوراثة، وهو من أقل فروع الطب دراسةً حاليًا، لا يستطيع تفسير ذلك حتى الآن.
الشيء الوحيد الممكن هو اتباع نهج مسؤول في التخطيط للحمل، والتسجيل، وإجراء الفحوصات في الوقت المناسب. ولكن، مرة أخرى، سيكون هذا الإجراء توعويًا أكثر منه وقائيًا، كأي فحوصات. لكن الآباء والأمهات الصغار سيعرفون مسبقًا ما يجب الاستعداد له، وفي حال الموافقة، سيقررون ما إذا كانوا قادرين على تحمل مسؤولية مثل تربية طفل مريض.
توقعات
يعتمد تشخيص متلازمة أنجلمان على طبيعة الخلل الكروموسومي وسرعة اكتشافه. الأطفال الأكثر تضررًا هم أولئك الذين يحتوي الكروموسوم 15 لديهم على "فجوات" في الجينات (الحذف). احتمالية قدرة هؤلاء المرضى على المشي والتحدث ضئيلة للغاية. يمكن تصحيح الحالات الأخرى باتباع نهج دقيق ورعاية طفلك.
للأسف، لن يتمكن هؤلاء المرضى من الاندماج الكامل في المجتمع، رغم أنهم ليسوا أغبياء، بل يفهمون الكلام ومعناه. ومع ذلك، سيعانون من مشاكل في التواصل طوال حياتهم. يمكن تعليم المرضى لغة الإشارة منذ الصغر، ولكن لا يمكن إجبارهم على التواصل بالكلمات. تقتصر مفردات المرضى "المتحدثين" على الحد الأدنى من الكلمات المستخدمة في الحياة اليومية (5-15 كلمة).
أما بالنسبة لمتوسط العمر المتوقع والصحة العامة لمرضى متلازمة أنجلمان، فتتراوح الأرقام هنا حول القيم المتوسطة. في مرحلة البلوغ، غالبًا ما يواجه المرضى مشاكل صحية مثل الجنف والسمنة، وهي مشاكل لا تُهدد الحياة باتباع نهج علاجي صحيح.